Palabras clave
INTRODUCCION
La miocardiopatía hipertrófica (MCH) es una enfermedad primaria del miocardio producida por mutaciones de genes que codifican proteínas del sarcómero1-7. Se considera que las mutaciones en el gen de la cadena pesada de la betamiosina (MYH7) causan entre un 15 y un 30% de los casos de MCH8-12, pero la frecuencia de mutaciones identificadas en MYH7 es variable en diferentes estudios y los datos disponibles en nuestro medio son muy escasos13,14. Por otra parte, el conocimiento sobre la correlación entre genotipo y fenotipo en las diferentes mutaciones descritas es muy limitado. El objetivo de este estudio es analizar la frecuencia de mutaciones en el gen de la cadena pesada de la betamiosina en una población amplia de pacientes con MCH y el análisis de la correlación genotipo-fenotipo en las mutaciones identificadas.
MÉTODOS
Sujetos del estudio
Se estudiaron 128 casos índice consecutivos con diagnóstico de MCH. Estos pacientes pertenecen a una cohorte de más de 400 pacientes con MCH controlada en una consulta específica de miocardiopatías de un hospital terciario que da servicio a una población de unos 500.000 habitantes. Prácticamente todos los pacientes diagnosticados de MCH en esta población son controlados en esta consulta que, además, realiza el seguimiento de pacientes remitidos de otros centros de la comunidad autónoma (2.500.000 habitantes). El diagnóstico de MCH se realizó de acuerdo con los criterios de la OMS y del grupo de trabajo de enfermedad miocárdica y pericárdica de la Sociedad Europea de Cardiología1,2. El estudio clínico en pacientes y familiares incluye: historia clínica y exploración física, electrocardiografía, ecocardiograma, Holter (en afectados), ergometría (en afectados), hemograma y bioquímica completa y recogida de muestras para extracción de ADN, plasma y suero. Todos los pacientes firmaron un consentimiento informado y el estudio fue aprobado por el comité de ética de la institución.
Análisis mutacional
A partir de muestras de sangre periférica se aisló el ADN mediante el kit de extracción de GFX Genomic Blood DNA Purification kit (Amersham Biosciences, Suecia)15. Se amplificó la secuencia codificante del gen de la cadena pesada de la betamiosina (MYH7), comprendida entre los exones 3 a 4016-18. Los primers se diseñaron desde las regiones intrónicas flanqueantes utilizando como secuencia de referencia la publicada en el GenBank con el número de acceso AJ238393. Los exones se analizaron mediante PCHM (polimorfismo en la conformación de hebras monocatenarias) en geles de poliacrilamida del kit GeneGel SSCP Starter Kit (Amersham Pharmacia Biotech, Suecia). Las muestras con patrón de movilidad anormal fueron reamplificadas, purificadas y secuenciadas. Un cambio en la secuencia de aminoácidos respecto a las secuencias de referencia16-19 se consideró como mutación patógena cuando cumplía los siguientes 3 criterios8,20: segregaba con los miembros afectados de la familia, no estaba presente en 200 cromosomas de individuos sanos no relacionados, y era un residuo conservado entre especies e isoformas de la miosina. Se consideraron como variantes alélicas raras los cambios que no segregaban con la enfermedad y que no estaban presentes en la población control. Se consideraron como polimorfismos no asociados con la MCH los cambios en la secuencia presentes en la población de control (100 individuos sanos, no emparentados entre sí). Las mutaciones fueron confirmadas independientemente mediante RFLP (restriction length fragment polymorphisms o polimorfismos de longitud de los fragmentos de restricción) o ARMS (amplified refractary mutation system o sistema de detección de mutaciones refractarias a la amplificación).
Estudio de la correlación genotipo-fenotipo
Se compararon las características demográficas, clínicas y ecocardiográficas en los casos índice con y sin mutación en el gen MYH7.
En los pacientes en los que se identificaron mutaciones se invitó a los familiares a realizar un estudio clínico, electrocardiograma (ECG), ecocardiograma y estudio genético, y se describen las características fenotípicas de los pacientes portadores de mutación en cada familia.
Análisis estadístico
Se utilizó el programa estadístico SPSS para PC versión 12.0. Se analizaron las diferencias entre pacientes con y sin mutación mediante pruebas exactas de la χ² para variables categóricas y mediante pruebas no paramétricas (U de Mann-Whitney) para variables continuas. En todos los análisis se consideraron estadísticamente significativas las diferencias en las que la p < 0,05 (contraste bilateral).
RESULTADOS
Mutaciones identificadas
Se hallaron 11 mutaciones (tabla 1), 7 de las cuales ya habían sido descritas previamente8,9,11,21-32. Estas mutaciones afectan a 13 de las 128 familias estudiadas (10,2%). La mutación I736T se identificó en 3 familias y la mutación A797T, en 2. En un paciente se identificaron 2 mutaciones: I736T y R787H.

Además, se identificó una variante alélica rara (R1781H, exón 37) que afecta a una residuo de la cola de la proteína, y 32 polimorfismos no asociados con MCH (presentes en la población de control), de los que sólo uno supone un cambio de aminoácido respecto a la secuencia de referencia (tabla 2).

Factores asociados con la presencia de mutaciones en el gen MYH7
En la tabla 3 se resumen las características de los pacientes con y sin mutación. Se identificó mutación en un 22% de los pacientes con antecedentes familiares de MCH y en un 33% de los que tenían antecedentes familiares de muerte súbita. Uno de cada 3 pacientes con grosor ≥ 30 mm presentó mutación. Los pacientes con mutación tenían hipertrofia más severa, menor diámetro telesistólico del ventrículo izquierdo y mayor fracción de eyección. No hubo diferencia significativa en la edad entre pacientes con y sin mutación, ni en el porcentaje de pacientes con hipertensión. Aunque la diferencia no alcanzó significación estadística, las mujeres presentaron un mayor porcentaje de mutaciones (tabla 3). No hubo diferencias significativas en cuanto a síntomas, tratamiento médico o intervenciones realizadas.

Correlación genotipo-fenotipo
En las tablas 4-7 se resumen los datos clínicos fundamentales en los casos índice y familiares de las 13 familias en las que se identificó alguna mutación. Disponemos de estudio genético en 42 miembros de las 13 familias, con resultado positivo en 29 (portadores de mutación) y negativo en 13. De los 29 pacientes con mutación, 6 (20%) correspondían a portadores sin criterios diagnósticos de MCH («portadores sanos»): 2 con la mutación I736T (16 y 21 años), uno con A797T (36 años) y 3 con K1459N (30, 33 y 34 años). Había antecedentes de muerte súbita en 9 familiares de 4 familias (4 muertes súbitas en 2 familias con I736T, uno con A797T, uno con R870H y 2 con A901P), y en ninguno de ellos se había realizado estudio genético.
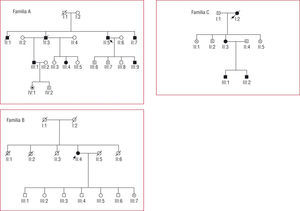
Mutación I736T (tabla 4; fig. 1)


Fig. 1. Árboles de las familias A, B y C, correspondientes a la tabla 4. Los cuadrados son varones y los círculos, mujeres. En negro, los pacientes con diagnóstico clínico de MCH. Los símbolos con N son sujetos sin MCH ni mutación. Los símbolos con punto negro central son portadores de mutación sin fenotipo de MCH. Los símbolos con una barra negra vertical indican sujetos con posible MCH por historia clínica (no comprobado). Los símbolos blancos son sujetos sin MCH conocida y no estudiados genéticamente. La línea diagonal indica sujetos fallecidos. Las flechas señalan los casos índice. MCH: miocardiopatía hipertrófica.
Se identificó en 3 familias con 8 portadores. La mayor parte de los portadores > 30 años presentaron hipertrofia severa, y en 2 casos se había realizado miectomía previa: uno requirió trasplante cardiaco por disfunción sistólica 13 años después y el otro falleció por insuficiencia cardiaca con 69 años. En las familias A y B hubo 4 muertes súbitas en varones entre 50 y 60 años sin diagnóstico previo.
Mutación R787H (tabla 4; fig. 1)
Se identificó en una mujer que además tiene la mutación I736T, con hipertrofia severa. Su madre había recibido una miectomía.
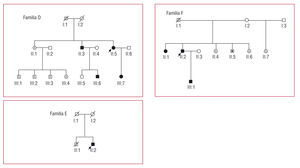
Mutaciones A797P y A797T (tabla 5; fig. 2)


Fig. 2. Árboles de las familias D, E y F, correspondientes a la tabla 5. Los cuadrados son varones y los círculos, mujeres. En negro los pacientes con diagnóstico clínico de MCH. Los símbolos con N son sujetos sin MCH ni mutación. Los símbolos con punto negro central son portadores de mutación sin fenotipo de MCH. Los símbolos con una barra negra vertical indican sujetos con posible MCH por historia clínica (no comprobado). Los símbolos blancos son sujetos sin MCH conocida y no estudiados genéticamente. La línea diagonal indica sujetos fallecidos. Las flechas señalan los casos índice. MCH: miocardiopatía hipertrófica.
La mutación A797T, previamente asociada con MCH, fue identificada en 2 familias. En los portadores el grado de expresión de la enfermedad es muy variable, con 2 portadores de 28 y 30 años con grosor parietal de 36 y 40 mm (los casos índice de las familias E y F), 2 portadores de 21 y 45 años con grosor de 14 y 15 mm y un portador de 36 años sin datos de MCH. En la familia E destaca el antecedente de una muerte súbita sin diagnóstico previo a los 22 años y en la F, una muerte por embolia a los 52.
La mutación A797P fue identificada en 4 pacientes de la familia D. En una portadora de 15 años, el ecocardiograma era normal pero el ECG era diagnóstico (ondas Q patológicas en cara anterolateral y voltaje alto del complejo QRS). En los demás portadores, la hipertrofia, de grado moderado, afectaba al septo y la pared anterior basal y media. El paciente de mayor edad presentaba dilatación de aurícula izquierda y anillo mitral, y requirió intervención quirúrgica por insuficiencia mitral severa sintomática con disfunción sistólica.
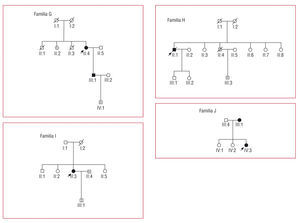
Mutación M388T (tabla 6; fig. 3)


Fig. 3. Árboles de las familias G, H, I y J, correspondientes a la tabla 6. Los cuadrados son varones y los círculos, mujeres. En negro los pacientes con diagnóstico clínico de MCH. Los símbolos con N son sujetos sin MCH ni mutación. Los símbolos con punto negro central son portadores de mutación sin fenotipo de MCH. Los símbolos con una barra negra vertical indican sujetos con posible MCH por historia clínica (no comprobado). Los símbolos blancos son sujetos sin MCH conocida y no estudiados genéticamente. La línea diagonal indica sujetos fallecidos. Las flechas señalan los casos índice. MCH: miocardiopatía hipertrófica.
El caso índice es una mujer con hipertrofia severa diagnosticada a los 54 años, a la que se implantó un desfibrilador por taquicardia ventricular monomorfa sostenida sincopal a los 62 años. Su hijo, deportista, presenta con 40 años una hipertrofia ligera con un ECG patológico (hipertrofia ventricular izquierda y ondas T negativas en la cara anterolateral).
Mutación G768R (tabla 6; fig. 3)
Identificada en un paciente con MCH no obstructiva con comportamiento restrictivo que había fallecido en coma hepático en lista de espera para trasplante cardiohepático.
Mutación R442C (tabla 6; fig. 3)
Se identificó en una mujer con hipertrofia localizada en la cara inferior (distribución atípica) y clínica de dolor torácico en relación con episodios de fibrilación auricular. Un hijo deportista presenta hipertrofia ligera concéntrica y ECG con ondas T negativas.
Mutación R663H (tabla 6; fig. 3)
Identificada en una mujer de 23 años con hipertrofia severa (42 mm) y obstrucción subaórtica severa. En el estudio familiar se diagnosticó MCH con hipertrofia septal ligera (15 mm) en su madre.
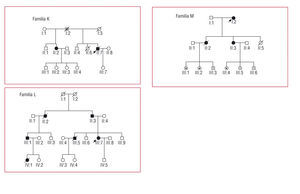
Mutaciones en el cuello y la cola de la proteína (tabla 7; fig. 4)


Fig. 4. Árboles de las familias K, L y M, correspondientes a la tabla 7. Los cuadrados son varones y los círculos, mujeres. En negro los pacientes con diagnóstico clínico de MCH. Los símbolos con N son sujetos sin MCH ni mutación. Los símbolos con punto negro central son portadores de mutación sin fenotipo de MCH. Los símbolos con una barra negra vertical indican sujetos con posible MCH por historia clínica (no comprobado). Los símbolos blancos son sujetos sin MCH conocida y no estudiados genéticamente. La línea diagonal indica sujetos fallecidos. Las flechas señalan los casos índice. MCH: miocardiopatía hipertrófica.
La mutación R870H se identificó en una mujer de 59 años con hipertrofia moderada no obstructiva y función sistólica conservada. Uno de sus hermanos tenía MCH no obstructiva con hipertrofia moderada que evolucionó durante un seguimiento de 12 años a disfunción sistólica severa, con adelgazamiento parietal y muerte súbita. La mutación A901P apareció en una mujer con hipertrofia severa (27 mm) con 2 muertes súbitas en la familia, con 15 y 45 años, y una muerte de causa no aclarada a los 35 años en una prima con MCH; además, tenía rachas de taquicardia ventricular no sostenida en el Holter y una respuesta anormal de presión arterial en la prueba de esfuerzo, por lo que se ha recomendado implante de desfibrilador. La mutación K1459N se identificó en 5 miembros de una familia, de los que 3 presentaron fenotipo normal (30, 31 y 33 años). El caso índice era una mujer diagnosticada con 76 años con grosor parietal de 27 mm. Una de sus hijas tenía MCH con hipertrofia ligera (15 mm) y estaba asintomática. Otra hija había fallecido con 53 años con clínica de insuficiencia cardiaca previa y diagnóstico de MCH.
DISCUSION
Estudios previos sugieren que las mutaciones en MYH7 causan entre un 15 y un 30% de los casos de miocardiopatía hipertrófica2,8,9. En nuestros pacientes, las mutaciones en este gen son menos frecuentes y aparecen en un 10% de las familias estudiadas. Esta diferencia puede tener diversas explicaciones. En primer lugar, la frecuencia de mutaciones depende del grado de selección de la población estudiada. Nuestro trabajo muestra que cuando se estudia a pacientes con hipertrofia severa, con antecedentes familiares de miocardiopatía hipertrófica (lo que implica una mayor penetrancia de las mutaciones o una manifestación clínica más precoz) o a pacientes con antecedentes familiares de muerte súbita, la frecuencia de mutaciones en este gen es superior y puede llegar hasta más del 30% de los casos. Por ello, es lógico que los estudios realizados en centros de referencia donde se remiten los pacientes con enfermedad más grave muestren mayor frecuencia de mutaciones. En este sentido, nuestra cohorte de pacientes representa a una población regional poco seleccionada, en la que la mayor parte de los pacientes proceden del ámbito del área sanitaria dependiente de nuestro hospital. En segundo lugar, aunque en este trabajo no hemos encontrado diferencias significativas en la edad en el momento del diagnóstico de pacientes con y sin mutación en el gen MYH7, la edad media de los pacientes con mutación era de 44 años, frente a 51 años en los pacientes sin mutación. Previamente se ha mostrado que la frecuencia de mutaciones en este gen es muy baja en los pacientes diagnosticados a edades avanzadas33, y debemos tener en cuenta que la edad media de diagnóstico en nuestros pacientes es unos 10 años superior a la de otras series. En tercer lugar, la frecuencia de mutaciones en diferentes genes puede variar en distintas poblaciones. En algunas zonas, la presencia de ciertas mutaciones con elevada frecuencia hace que la proporción de mutaciones identificadas en otros genes sea menor34,35.
Un dato interesante de nuestro estudio es la mayor frecuencia de mutaciones identificadas en las mujeres. El patrón de herencia habitual en la MCH es autosómico dominante y cabría esperar que un 50% de los pacientes fueran mujeres. Sin embargo, en nuestra población y en prácticamente todas las series descritas, la proporción de mujeres está entre un 30 y un 40%, con una mayor sintomatología de la enfermedad en las mujeres36. La identificación de mayor número de mutaciones en pacientes de sexo femenino junto con el dato de que estas mutaciones aparecen con mayor frecuencia en pacientes con hipertrofia severa confirma que la diferencia en la prevalencia de MCH entre varones y mujeres se debe a una menor expresión morfológica de la enfermedad en el sexo femenino.
Actualmente, muchos autores insisten en el concepto de que la mayor parte de las mutaciones identificadas en pacientes con MCH son «mutaciones privadas» o nuevas mutaciones. Nuestro trabajo, en el que se ha realizado un estudio sistemático del gen MYH7, confirma que un elevado porcentaje de las mutaciones que se identifican actualmente ha sido ya previamente descrito (7 de 11 en nuestro caso). Por otra parte, 2 de las mutaciones identificadas aparecieron en varias familias que inicialmente no se consideraban relacionadas. La identificación de mutaciones en diferentes familias permite hacer una valoración más precisa de la correlación genotipo-fenotipo y la interpretación adecuada del papel patogénico de cada mutación. Varios hallazgos de nuestro estudio inciden en la importancia de realizar un estudio familiar completo. Mientras que en algunas mutaciones, como la I736T, el fenotipo se reproduce de forma similar en la mayor parte de los portadores, en otras, como la A797T o la R663H, llama la atención la gran diferencia entre el fenotipo de los casos índice (hipertrofia severa en pacientes jóvenes) y los familiares portadores con hipertrofia ligera, a pesar de ser de edades similares o mayores. En estos casos se debe considerar la posibilidad de que haya factores genéticos o ambientales adicionales que expliquen la gran diferencia de expresión. En varios trabajos se ha demostrado que los pacientes con MCH pueden presentar más de una mutación y que la presencia de dobles mutaciones se asocia con una expresión más severa de la enfermedad8. En nuestro estudio, que se ha centrado sólo en el gen MYH7, hemos identificado la presencia de 2 mutaciones previamente asociadas con el desarrollo de MCH en un caso, por lo que no sería raro que otros pacientes pudieran tener mutaciones adicionales en otros genes.
Gran parte de los estudios sobre la presencia de mutaciones en el gen MYH7 han limitado el análisis a las regiones que codifican la cabeza y el cuello de la betamiosina. En estudios más recientes se ha demostrado que también mutaciones que afectan a la cola de la proteína pueden asociarse con la enfermedad. Nosotros identificamos una mutación que afecta a la cola de la betamiosina (K1459N). Esta mutación no afecta a la probabilidad de formar hélices superenrolladas de la cola de la miosina, lo que puede explicar un fenotipo benigno con penetrancia incompleta y expresión tardía. De las mutaciones identificadas en la región del cuello de la proteína destaca la mutación A901P, no descrita previamente. Se observó que no estaba presente en 200 cromosomas de individuos control, por lo que se descartó que fuese una variante presente en la población. La estructura de hélice alfa superenrollada que caracteriza a la región del cuello de la MYH7 se ve completamente alterada por la presencia de una prolina, ya que es un residuo incompatible con este tipo de estructuras. Esta seria alteración en la estructura de la proteína puede explicar en parte la severidad del fenotipo en la familia estudiada, con múltiples muertes súbitas.
En las 13 familias estudiadas se ha registrado un total de 8 muertes súbitas. Sólo en uno de los casos conocemos el diagnóstico genético (portador obligado). Esta falta de evidencia sobre el diagnóstico genético en sujetos que han fallecido súbitamente es habitual en los estudios publicados y representa una limitación importante a la hora de interpretar el significado pronóstico de las diferentes mutaciones identificadas en sus familias. Es importante tener en cuenta que los pacientes que mueren súbitamente podrían tener características diferenciales como, por ejemplo, mutaciones adicionales. La recogida sistemática de muestras para estudio genético en pacientes con MCH, su seguimiento clínico y el estudio familiar lo más completo posible son fundamentales para llegar en el futuro a conclusiones sólidas sobre las implicaciones pronósticas de las diferentes mutaciones.
Limitaciones
El estudio se realizó mediante PCHM y secuenciación de los fragmentos con motilidad anormal. La sensibilidad de la PCHM es de un 84-89%37,38, por lo que podría haber alguna mutación adicional que no hubiera sido identificada.
Aunque se ha realizado un estudio lo más amplio posible en las familias de los pacientes con mutación, éste no ha sido completo. La recogida de muestras no es posible en sujetos fallecidos ni en los que han declinado participar en el estudio o no han sido avisados por el caso índice.
El estudio se ha centrado en el análisis del gen MYH7. La posible influencia en el fenotipo de mutaciones en otros genes relacionados con el desarrollo de MCH no ha podido ser evaluada.
En conclusión, las mutaciones en el gen MYH7 son una causa relativamente frecuente de MCH en nuestro medio. La probabilidad de identificar mutaciones en este gen es mayor en pacientes con antecedentes familiares de muerte súbita y con hipertrofia severa. La mayor parte de las mutaciones identificadas había sido descrita previamente en otras poblaciones y algunas se repiten en varias familias. Por otro lado, nuestro trabajo ha identificado 4 nuevas mutaciones asociadas con la enfermedad. Ciertas mutaciones muestran una correlación genotipo-fenotipo relativamente estable, mientras que en otras, las marcadas diferencias entre el fenotipo de los casos índice y sus familiares hacen sospechar la presencia de factores genéticos adicionales que debemos identificar. Los estudios genéticos son fundamentales para entender la gran heterogeneidad clínica y pronóstica de la MCH. Para que la genética sea una herramienta útil en la toma de decisiones clínicas es necesario que dispongamos de información detallada sobre las características clínicas y morfológicas de los portadores de las diferentes mutaciones, como la que proporciona este estudio.
AGRADECIMIENTOS
Agradecemos a las enfermera Elena Veira su trabajo en la recogida de muestras y datos de pacientes y familiares. Nuestro agradecimiento también para los pacientes y sus familias por su participación y colaboración desinteresada.
Véase editorial en págs. 994-6
Este estudio ha sido financiado con una beca de la Sociedad Española de Cardiología y Fundación Española de Corazón para investigación básica en cardiología y con fondos de investigación de la Red de Investigación Cardiovascular RECAVA del Instituto de Salud Carlos III. Lorenzo Monserrat recibe financiación de una ayuda a la investigación de la Fundación Sanofi-Aventis. Carlos Dumont recibe financiación de BBVA-Fundación Carolina.
Correspondencia: Dr. L. Monserrat Iglesias.
Servicio de Cardiología. CHU Juan Canalejo.
As Xubias, 84. 15006 A Coruña. España.
Correo electrónico: lorenzo_monserrat@canalejo.org
Recibido el 19 de abril de 2006.
Aceptado para su publicación el 27 de junio de 2006.