En la última década, la proteómica y la metabolómica han realizado aportaciones sustanciales al conocimiento de las enfermedades cardiovasculares. La evaluación no sesgada de los procesos fisiopatológicos sin partir de presunciones establecidas a priori complementa otras técnicas de biología molecular que se emplean en la actualidad con un enfoque reduccionista. En la presente revisión, se resaltan algunos de los métodos de las ciencias «ómicas» que se emplean para evaluar los cambios de las proteínas y los metabolitos en la enfermedad cardiovascular. Es muy infrecuente que una función biológica específica discreta se atribuya a una sola molécula; es más habitual que se lleve a cabo con la aportación combinada de muchas proteínas. A diferencia del enfoque reduccionista, en el que se estudia individualmente las moléculas, las plataformas «ómicas» permiten el estudio de interacciones más complejas en sistemas biológicos. La combinación de la proteómica y la metabolómica para cuantificar los cambios de los metabolitos y sus correspondientes enzimas hará avanzar nuestro conocimiento de los mecanismos fisiopatológicos y facilitará la identificación de nuevos biomarcadores de enfermedad cardiovascular.
Palabras clave
La enfermedad cardiovascular (ECV) es la primera causa de mortalidad y morbilidad en los países industrializados. La predicción de los eventos cardiovasculares se basa en el seguimiento de los factores de riesgo convencionales, como edad, sexo, hábito tabáquico, diabetes mellitus e hipertensión1. Muchos de estos factores de riesgo tienen una prevalencia elevada en la población, e incluso los mejores algoritmos para los eventos coronarios agudos han fallado en la predicción de la mayoría de los casos de ECV en un periodo de 10 años2. Además de las dificultades de predicción, las ECV, como la insuficiencia cardiaca, pueden constituir un espectro heterogéneo de etiologías, estadios patológicos y bases genéticas. Hay una urgente necesidad de identificar nuevos biomarcadores para estratificar a los pacientes y personalizar los tratamientos. Actualmente, la evaluación de biomarcadores se basa en la cuantificación de unas pocas proteínas o metabolitos3. Las plataformas de alto rendimiento como la proteómica y la metabolómica pueden ofrecer lecturas simultáneas de centenares de proteínas y metabolitos. En esta revisión, se resumen las plataformas de proteómica y metabolómica que hoy se aplican a la investigación cardiovascular y pueden conducir a la identificación de nuevos biomarcadores de utilidad clínica.
PROTEÓMICALa primera secuencia completa del genoma humano se publicó en 2001. En contra de lo esperado, esta secuencia incluía tan solo unos 20.000 a 25.000 marcos de lectura abiertos que codificaban proteínas4. Sin embargo, los productos génicos están sujetos a procesos de corte y empalme y edición de ARN alternativos que dan lugar a una amplia variedad de isoformas diferentes de las proteínas5. El objetivo de la proteómica es interrogar al proteoma6. El proteoma engloba todo el conjunto de proteínas expresadas por una célula, un tejido o un organismo, incluidas sus modificaciones postraduccionales. La proteómica es el análisis integral de la expresión génica con el empleo de diversas técnicas para identificar y caracterizar las proteínas. El término proteómica fue acuñado por Marc Wilkins en 1994 por analogía con el de la genómica, que es el análisis de los genes.
Las primeras técnicas de proteómica se desarrollaron en la década de los setenta7 y su uso ha evolucionado continuamente desde entonces. No fue hasta mediados de la década de los noventa cuando se aplicó la proteómica al estudio de las ECV, con los trabajos pioneros de Knecht et al8 y Jungblut et al9. En esa época, una de las dificultades principales era la identificación de las proteínas. Inicialmente, se utilizó la secuenciación de Edman, pero esta técnica ha sido sustituida tras su introducción por la espectrometría de masas (EM) biológica. El primer Premio Nobel motivado por la EM fue el concedido a F.W. Aston en 1920. Su espectrómetro de masas permitía la separación de diferentes isótopos. Más recientemente, dos nuevos inventos han hecho posible analizar biomoléculas (ADN, péptidos, proteínas) mediante EM: en 1987, M. Karas y F. Hillenkamp inventaron la MALDI (desorción/ionización con láser asistida por matriz). En la MALDI-EM, se mezcla una matriz (p. ej., ácido α-ciano-4-hidroxicinnámico) con el producto a analizar (p. ej., péptidos). Dicho producto es desorbido de la matriz con la aplicación de un láser y es ionizado10. En 1989, J.B. Fenn inventó la ionización por electrospray11, lo que le valió el Premio Nobel de Química en 2002. En la ionización por electrospray, el producto analizado se ioniza de una fase líquida a otra gaseosa. Así se puede establecer una interfase directa de los sistemas de cromatografía líquida (CL) con los espectrómetros de masas. La CL-EM en tándem (CL-EM/EM) es el patrón de referencia actual de la proteómica. La CL separa primero los péptidos, lo cual es esencial pues la mayor parte de las mezclas son demasiado complejas para analizarlas mediante EM sin prefracionamiento. A continuación, el espectrómetro de masas en tándem registra las masas de los péptidos inalterados (EM plena) antes de seleccionar un ion precursor y fragmentarlo. Habitualmente la fragmentación se induce mediante colisión con argón o nitrógeno. Los métodos más recientes emplean también electrones, un método de fragmentación más suave que preserva las modificaciones postraduccionales12. Los fragmentos se registran en un espectro de EM/EM y el patrón de fragmentación revela un Δ masa específico para cada aminoácido presente en el péptido. En los estudios iniciales de proteómica, el proceso de trabajo se basaba en un paso inicial de separación proteica que utilizaba técnicas como la electroforesis en gel. A medida que se fueron desarrollando espectrómetros de masas más rápidos y más sensibles, se efectuaban la digestión y el análisis directo de mezclas de proteínas cada vez más complejas, sin un fraccionamiento previo de proteínas. Se denomina a este método proteómica de abajo arriba. En cambio, la proteómica de arriba abajo analiza proteínas inalteradas mediante EM. Sin embargo, este método continúa estando limitado a una sola proteína en soluciones.
Espectrometría de masas en tándemLas técnicas de abajo arriba hoy son las utilizadas para la mayor parte de la labor de análisis de muestras biológicas: en la «proteómica de perdigonada» se analizan las proteínas de mezclas complejas utilizando una combinación de CL de alto rendimiento y EM/EM. Una posible advertencia respecto a esta técnica es que el espectrómetro de masas selecciona el ion precursor más abundante para la fragmentación. Por consiguiente, la detección de las proteínas abundantes es más probable que la de las escasas. El muestreo insuficiente es especialmente problemático en el caso de muestras como las de plasma o suero, que corresponden al proteoma más complejo del cuerpo humano, con proteínas que son de entre 10 y 12 órdenes de magnitud en el rango dinámico lineal13. Los espectrómetros de masas actuales solamente resuelven de 4 a 5 órdenes de magnitud. Aunque un solo péptido puede ser suficiente para identificar sin ambigüedad una proteína, son necesarios múltiples péptidos de la misma proteína para una cuantificación fiable en la proteómica de perdigonada. Hasta la fecha, la mayoría de los estudios de proteómica que han analizado muestras de plasma han fracasado en el intento de identificar nuevos biomarcadores debido a que las proteínas poco abundantes son difíciles de detectar en presencia de componentes muy abundantes como la albúmina. El problema del muestreo insuficiente del proteoma plasmático se podría superar en parte con técnicas que utilizan una depleción de las proteínas plasmáticas abundantes. Una estrategia alternativa es el empleo de tejido enfermo, en el que hay un enriquecimiento de biomarcadores. Puede usarse una estrategia más específica en el plasma/suero una vez identificado el biomarcador candidato.
Monitorización por reacción múltipleLa monitorización por reacción múltiple en un espectrómetro de masas de triple-cuadrupolo (QqQ-EM) se aplica a péptidos o metabolitos identificados en los experimentos de descubrimiento14,15. Una QqQ-EM proporciona una forma efectiva y exacta de cuantificar unas pocas moléculas diana seleccionadas, incluso en mezclas complejas como el plasma o el suero15. Si se puede ionizar la molécula de interés mediante ionización por electrospray, la QqQ-EM tendrá una interfase con un sistema de CL para la separación del producto analizado, tal como se ha mencionado antes. La molécula precursora se selecciona entonces en el primer cuadrupolo. El segundo cuadrupolo se emplea como cámara de colisión para la fragmentación. Los iones producidos son detectados en el tercer cuadrupolo, y su intensidad es un indicador de la abundancia de la molécula de origen16. Dada su alta especificidad, la monitorización por reacción múltiple se emplea para la validación de biomarcadores candidatos identificados en los experimentos de descubrimiento17, pero se ha empleado también con éxito para confirmar lugares específicos de fragmentación de las proteínas18. Además, la QqQ-EM es el método de elección para los estudios de metabolómica dirigida empleando metabolitos estándares como calibradores19.
METABOLÓMICALos metabolitos son los productos finales de todos los procesos que se producen en las células, y las cantidades de metabolitos en la enfermedad reflejan la adaptación de los sistemas biológicos a los estados patológicos. Hoy se estima que hay más de 2.000 metabolitos diferentes que pueden ser sintetizados de forma endógena20. Además, la dieta incorpora metabolitos exógenos, como por ejemplo las vitaminas. Otro factor contribuyente importante para el conjunto de metabolitos del organismo es la flora intestinal. Al igual que en el caso de la proteómica, el objetivo de la metabolómica es caracterizar el complemento de moléculas pequeñas de una muestra determinada e interrogar a las redes metabólicas en condiciones normales y patológicas, de manera cualitativa y cuantitativa. Las tecnologías de metabolómica se han aplicado a diferentes áreas de investigación clínica, entre ellas el descubrimiento de biomarcadores y fármacos21,22, la toxicología23 y la nutrición24. Los primeros estudios de metabolómica se publicaron a comienzos de la primera década de este siglo y se aplicaron rápidamente a la investigación cardiovascular. Al igual que en la proteómica, la EM es el método de elección para los análisis de metabolitos, pero también se emplea ampliamente la espectroscopia por resonancia magnética (ERM)25.
Metabolómica basada en espectroscopia por resonancia magnéticaLa ERM es menos sensible que la EM, pero es notablemente cuantitativa y aporta ventajas en la cuantificación de los metabolitos en tejidos intactos y en extractos de tejidos26. Esta técnica se basa en que ciertos núcleos con un número de nucleones (protones o neutrones) impar poseen una propiedad denominada espín nuclear. Las resonancias de los núcleos excitados mediante radiofrecuencias específicas pueden medirse para producir una señal bien definida. Los protones (1H) son con frecuencia los núcleos de elección para el parámetro de espín, ya que son abundantes. Empleando ácido perclórico, se puede extraer los metabolitos hidrosolubles directamente de corazones sometidos a congelación brusca. La 1H-ERM proporciona una forma cuantitativa de medir los metabolitos hidrosolubles presentes en estos extractos de metabolitos27. Se utiliza un patrón de referencia interno para calibrar las desviaciones químicas en los espectros para las identificaciones de los metabolitos. Es importante señalar que el área del pico que corresponde a la señal de cada metabolito puede calcularse mediante la comparación con el patrón de referencia interno para obtener valores cuantitativos. Pueden examinarse al mismo tiempo muchas clases de moléculas en las muestras extraídas, sin partir de supuestos previos respecto a los tipos de moléculas. Esto hace que la 1H-ERM sea un método excelente para abordar los análisis no dirigidos. La 1H-ERM muestra su potencia a la sensibilidad requerida principalmente en extractos ácidos. La sensibilidad y la resolución se reducen en los tejidos sólidos. Se puede analizar los tejidos sólidos empleando una técnica denominada 1H-ERM con spinning de ángulo mágico de alta resolución. El estado energético de una célula o un tejido determinados se puede evaluar con ERM de fósforo (31P-ERM). Esta permite la detección de metabolitos energéticos cardiacos de vida corta como fosfocreatina y adenosintrifosfato, así como fosfato inorgánico y el pH intracelular. La 31P-ERM también es adecuada para determinaciones in vivo e in vitro28,29.
Metabolómica basada en espectrometría de masasLa metabolómica basada en EM proporciona un método muy sensible de recopilar la información de un perfil metabólico, especialmente en los líquidos corporales30. Se han realizado análisis metabolómicos basados en el plasma después de una isquemia miocárdica31, pruebas de esfuerzo y pacientes prediabéticos32. Los metabolitos se separan antes de la identificación mediante espectrofotometría de masas, utilizando para ello cromatografía de gases o CL. Al igual que en la proteómica, los avances recientes en las tecnologías de EM han transformado nuestra capacidad de establecer un perfil de metabolitos no solo en solución, sino también directamente a partir de cortes de tejido. Por ejemplo, Manicke et al33 aplicaron una EM con ionización por electrospray de desorción para visualizar e identificar especies moleculares de lípidos en placas de origen humano. En otro estudio se mostró el uso de técnicas de imagen de moléculas pequeñas mediante EM en tejido cardiaco de ratón34. Nosotros utilizamos los avances más recientes de la EM en lipidómica de perdigonada para comparar el contenido lipídico de lesiones de aterosclerosis humanas bien definidas. Comparando las muestras de endarterectomía de pacientes sintomáticos y asintomáticos y áreas estables e inestables de la misma lesión sintomática, delimitamos las firmas de lípidos características para determinar la vulnerabilidad de la placa18.
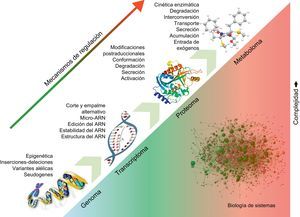
APLICACIONES A LA ENFERMEDAD CARDIOVASCULARConocimiento del mecanismo de la enfermedadA diferencia del genoma, el proteoma y el metaboloma son dinámicos y están mucho más próximos al fenotipo de la enfermedad (Fig.). Así pues, podrían conducir a un mejor conocimiento de los procesos que causan que la ECV se manifieste y progrese. Actualmente, las técnicas de proteómica pueden identificar hasta 5.000 proteínas en una muestra biológica/clínica compleja, pero las plataformas de proteómica están en continua evolución y amplían las fronteras de la instrumentación analítica35. En la investigación cardiovascular, el rango dinámico de la expresión proteica a que dan lugar las proteínas de miofilamentos abundantes (p. ej., tropomiosinas, miosinas, titina) dificulta la detección de las proteínas poco abundantes en los miocardiocitos. En consecuencia, los investigadores han desarrollado métodos analíticos dirigidos a subproteomas específicos, como los de los miofilamentos cardiacos36, las mitocondrias cardiacas37, las membranas celulares38 o el núcleo celular39. Nosotros hemos caracterizado recientemente el remodelado de la matriz extracelular vascular en aneurismas de aorta abdominal humanos, así como la fibrosis inicial y tardía, en un modelo porcino de isquemia/lesión de reperfusión40,41. Con la determinación de los niveles de expresión de proteínas sin partir de supuestos establecidos a priori, las tecnologías avanzadas pueden generar nuevas hipótesis y desplazar el centro de interés actual de la determinación cuantitativa a las diferencias cualitativas en la fibrosis cardiaca que pueden modificar la progresión de la enfermedad. Las técnicas «ómicas» abordan las interacciones complejas que se dan dentro de los sistemas biológicos desde un punto de vista holístico, en particular las interacciones que afectan a la fisiopatología de la enfermedad. Anteriormente, los enfoques reduccionistas convencionales trataban las cascadas de señalización intracelulares como modelos lineales, considerando las moléculas involucradas confinadas a una única vía de señal. Sin embargo, las diferentes vías interactúan entre sí y están organizadas en forma de redes que incluyen tanto proteínas como moléculas pequeñas42. La bioinformática ha pasado a ser un instrumento esencial para analizar y visualizar de manera completa estas interacciones, y bases de datos como la KEGG (Kyoto Encyclopedia of Genes and Genomes) o Reactome establecen mapas de enzimas/metabolitos regulados dentro de diferentes redes metabólicas43. Así pues, la combinación de proteómica y metabolómica proporciona una lectura complementaria que mejora la fiabilidad de la interpretación de los datos44 y ofrece un conjunto de instrumentos no sesgados para interrogar al metabolismo cardiovascular45,46.

Biología de sistemas. Los procesos reguladores en el ADN afectan a la expresión de moléculas de procesos posteriores, como los ARN, las proteínas y los metabolitos. Los efectos de los diferentes elementos reguladores son aditivos. La biología de sistemas intenta analizar las interacciones entre las diferentes entidades moleculares para ofrecer una visión holística de los procesos biológicos y las alteraciones patológicas que se producen en la enfermedad.
La fibrilación auricular (FA) conduce a varias formas diferentes de remodelado auricular, denominadas eléctrico, contráctil y estructural. Empleando técnicas de proteómica y metabolómica para analizar la muestra de orejuela auricular derecha de pacientes en ritmo sinusal y FA permanente, observamos una posible nueva forma de remodelado auricular-remodelado metabólico47. Varias enzimas involucradas en el metabolismo de la glucosa, los lípidos y la energía fueron alterados durante la FA. Los resultados proteómicos se sustanciaron mediante análisis metabolómico, y revelaron un aumento de los metabolitos lipídicos y los cuerpos cetónicos (acetato, betahidroxibutirato). No se conoce la contribución del remodelado metabólico a la FA que se hace persistente. Sin embargo, las alteraciones metabólicas parecen preceder a la aparición de la FA, lo cual sugiere que no son únicamente una consecuencia de esta. En orejuelas auriculares humanas procedentes de pacientes con o sin FA postoperatoria, se observó que había desregulación de metabolitos cardiacos y alteración de las concentraciones de enzimas relacionadas con el metabolismo de la energía antes del inicio de las arritmias postoperatorias47. De manera análoga, las alteraciones proteómicas y metabolómicas observadas en un modelo animal de un sustrato de FA en evolución (marcapasos taquicárdico ventricular en el perro) señalan la importancia clave de los cambios metabólicos, el estrés oxidativo y la lesión estructural asociada a la insuficiencia cardiaca congestiva y las alteraciones profibrilatorias en la aurícula izquierda48.
Identificación de biomarcadores candidatosEn la segunda mitad del siglo xx hemos asistido a la generalización de las intervenciones quirúrgicas mínimamente invasivas para las ECV, así como a una aplicación más amplia de diversas moléculas farmacológicas, como estatinas, bloqueadores beta y tratamientos antiagregantes plaquetarios y anticoagulantes, que han llevado a una enorme mejora en los resultados clínicos y el pronóstico de los pacientes con ECV. Al mismo tiempo, la generalización de las pruebas estándares no invasivas y rápidas para detección precoz o valoración de los factores de riesgo ha mejorado ciertamente el diagnóstico y los tratamientos preventivos. Sin embargo, las ECV rara vez se atribuyen a un único factor. En los últimos años, se ha puesto de manifiesto la necesidad de nuevos biomarcadores para las ECV, con objeto de estratificar mejor a los pacientes y determinar su respuesta al tratamiento. Hoy se considera a los biomarcadores entidades únicas. En una época de tecnologías de alto rendimiento, han aparecido nuevos enfoques para abordar múltiples biomarcadores en el diagnóstico clínico y el pronóstico. Por ejemplo, los espectrómetros de masas están ya en el uso clínico para el diagnóstico perinatal y toxicología. Mientras que los inmunoanálisis y los métodos bioquímicos han dominado el campo de los análisis clínicos, las técnicas de alto rendimiento podrían pasar a utilizarse más ampliamente para los biomarcadores de la ECV en los próximos años.
CONCLUSIONESEl uso combinado de proteómica y metabolómica, junto con los nuevos métodos para el análisis de datos, permite obtener una imagen más holística de los sistemas biológicos y sus alteraciones en la enfermedad. En los sistemas biológicos, las moléculas (ARN, proteínas o metabolitos) interaccionan para llevar a cabo los procesos moleculares. No se debe abordar estas complejas interacciones exclusivamente con un enfoque reduccionista. La ventaja de las técnicas «ómicas» es que se puede usarlas para comparaciones de muestras clínicas no sujetas a ninguna hipótesis. Esto ha sido posible principalmente gracias a tres factores clave: a) las mejoras alcanzadas en la sensibilidad y la exactitud de los espectrómetros de masas; b) el desarrollo de mejores técnicas de separación, junto con nuevos métodos de marcado, y c) la disponibilidad de bases de datos para analizar series de datos de complejidad creciente. Los espectrómetros de masas serán la tecnología clave en la proteómica y la metabolómica para hacer avanzar nuestro conocimiento de las ECV y aportar nuevos biomarcadores candidatos49.
CONFLICTO DE INTERESESNinguno.
El Prof. Mayr es miembro pleno de la British Heart Foundation. La investigación contó con el apoyo del NIHR (National Institute for Health Research), el Biomedical Research Centre del Guy's and St. Thomas’ NHS Foundation Trust y el King's College London, en colaboración con el King's College Hospital.