
Los niveles del microRNA-16-5p (miR-16) aumentan en la cardiopatía isquémica y modelos animales de miocardiopatía dilatada isquémica (MCDi) vinculándolo con la apoptosis. Proponemos determinar el papel del miR-16 en la respuesta adaptativa celular asociada al estrés del retículo endoplasmático (RE) y la autofagia en el entorno MCDi.
MétodosSe cuantificaron los niveles plasmáticos del miR-16 en 168 participantes (n=76 controles, n=60 MCDi y n=32 MCD familiar [portadores de la variante patogénica BAG3]), mediante la Reacción en Cadena de la Polimerasa cuantitativa a Tiempo Real, correlacionándose con las variables clínicas. La influencia de la sobreexpresión del miR-16 se examinó en una línea celular cardiaca humana. Se analizó la apoptosis, viabilidad celular y los niveles de biomarcadores del estrés del RE, daño cardíaco y autofagia.
ResultadosMiR-16 está sobreexpresado en el plasma de MCDi (p=0,039). El modelo de regresión logística multivariante relacionó los niveles de miR-16 con varias variables clínicas de la MCDi (p <0,001). In vitro, la sobrexpresión del miR-16 induce apoptosis (p=0,02), disminuyendo la viabilidad celular (p=0,008). MiR-16 promueve un papel pro-apoptótico del RE mediado por la vía PERK/CHOP. Sin embargo, observamos la inducción de la autofagia por miR-16 (p <0,0001) sin bloquearse, como posible mecanismo citoprotector.
ConclusionesEl miR-16 está específicamente relacionado con la MCDi. En un entorno isquémico, miR-16 induce el estrés del RE promoviendo la inflamación seguida de la autofagia, como respuesta a la homeostasis celular en células cardiacas.
Palabras clave
La insuficiencia cardiaca (IC) ha alcanzado proporciones de pandemia mundial y se asocia con elevadas morbilidad y mortalidad1. Una causa frecuente de la IC es la miocardiopatía dilatada (MCD)1. La MCD se define por dilatación del ventrículo izquierdo (VI) o biventricular y un deterioro de la contracción que no se explica por unas condiciones de carga anormales (es decir, hipertensión y valvulopatía) o por enfermedad coronaria2. La MCD isquémica (MCDi) es la etiología de MCD más frecuente3. Afecta a pacientes con antecedentes de infarto agudo de miocardio o revascularización o se origina por enfermedad coronaria4.
La fisiopatología de la MCDi a escala subcelular continúa sin esclarecerse5. Después de un episodio de isquemia, la apoptosis desempeña un papel crucial en la IC6. En la homeostasis de los cardiomiocitos intervienen múltiples procesos celulares, como el estrés del retículo endoplásmico (RE) y la autofagia7. La señalización del estrés del RE tiene un efecto citoprotector e intenta mantener esta homeostasis, pero un estrés del RE prolongado da lugar a disfunción de los cardiomiocitos y apoptosis7. Por otro lado, se han identificado vacuolas de autofagia en el VI de pacientes con MCD8,9. Sin embargo, es necesario un conocimiento más profundo de la relación existente entre el estrés del RE y la autofagia por lo que respecta a las enfermedades cardiovasculares.
Los microRNA (miRNA) circulantes han adquirido importancia como biomarcadores diagnósticos y como diana para el tratamiento de la MCD (y ello ha sido objeto de una revisión detallada en otra publicación)3. Los miRNA desempeñan un papel clave en las vías de señalización subyacentes a la fisiopatología de varias enfermedades cardiovasculares, incluida la MCD. Por ejemplo, el microRNA-16-5p (miR-16) y el miR-27 muestran una asociación positiva con el deterioro del VI después de un infarto agudo de miocardio10. Además, las concentraciones circulantes e intracelulares de miR-16 están aumentadas en los modelos de la MCDi de rata11,12 y también en el corazón fetal humano13. La sobrexpresión del miR-16 fomenta la apoptosis del miocardio a través de una regulación de la expresión de la proteína del linfoma de linfocitos B 2 (BCL-2) en el modelo de isquemia de rata12. En el mismo modelo, el knockdown del miR-16 tiene un efecto cardioprotector11. Sin embargo, no se ha definido aún la influencia del miR-16 en los mecanismos intracelulares en la población humana con MCDi.
El objetivo del estudio es determinar el papel del miR-16 en la respuesta de adaptación celular asociada con el estrés del RE y la autofagia en unas condiciones de MCDi apoptótica.
MÉTODOSPoblación del estudio y obtención de muestras de sangreEn este estudio de casos y controles, se incluyó a un total de 76 controles, 60 pacientes con MCDi y 32 pacientes con MCD familiar (con una variante patogénica del BAG3). Se consideró aptos para el estudio solo a mayores de 18 años. Se obtuvo una información clínica y farmacológica detallada de cada uno de los participantes. La MCDi de todos los pacientes con MCDi se confirmó mediante cateterismo coronario, según lo recomendado por la guía de la Sociedad Europea de Cardiología2. Se obtuvieron muestras de plasma según lo descrito anteriormente14–16. Nuestro protocolo fue aprobado por el Comité de Ética de Investigación de Cádiz (España). Todos los pacientes dieron su consentimiento informado por escrito. Este estudio se llevó a cabo cumpliendo los principios éticos de la Declaración de Helsinki.
Cuantificación de los miRNA circulantesSe extrajo el ARN total con el kit avanzado miRNeasy para suero/plasma (Qiagen, Alemania). Las concentraciones de miR-16 circulantes se determinaron mediante reacción en cadena de la polimerasa en tiempo real cuantitativa (qRT-PCR). Se sintetizó el cDNA específico del miRNA con el kit de miRCURY RT LNA (Qiagen). La qRT-PCR se llevó a cabo con el kit de miRCURY PCR SYBR Green (Qiagen) y las concentraciones de miR-16 se normalizaron con el método 2−ΔCq.
Cultivo celular y transfecciónSe cultivó la línea celular AC16 humana, que desarrolla muchas de las propiedades bioquímicas de las células cardiacas, según lo descrito anteriormente17. Se aplicó a las células una transfección con 50pmol de hsa-miR-16-5p (miR-16) y con mimetizadores positivo miRNA mirVana (miR-1) y negativo (control negativo n.o 1) utilizando Lipofectamine RNAiMAX y siguiendo las recomendaciones del fabricante (Thermo Fisher Scientific, Estados Unidos) durante 6, 16, 24 y 48 h. Para medir el flujo autofágico, las células se trataron con 2 compuestos de control de autofagia —cloroquina 120 μM y rapamicina 500 nM (Sigma-Aldrich, Estados Unidos)— durante 16 y 2 h respectivamente.
Aislamiento del ARN total y reacción en cadena de la polimerasa en tiempo real cuantitativa en células AC16Se aisló ARN de células AC16 con el kit de aislamiento de ARN MagMAX (Thermo Fisher Scientific). Se realizó una transcripción inversa del ARN con el kit de transcripción inversa de cDNA de alta capacidad (Thermo Fisher Scientific) siguiendo las instrucciones del fabricante. El análisis de la expresión génica de miR-16, ATG14, TWF1, U6 snRNA y GAPDH se realizó con un test de expresión génica TaqMan (Applied Biosystems, Estados Unidos). La cuantificación relativa de las concentraciones de mRNA (los pares de cebador se muestran en la tabla 1) se realizó con 6,5 ng de cDNA, cebadores directos e inversos a concentración de 100nM cada uno y un kit SYBR Green PCR Master Mix Reagent (Life Technologies, Estados Unidos). Las concentraciones de mRNA se normalizaron respecto a las de GAPDH.
Oligonucleótidos para la reacción en cadena de la polimerasa en tiempo real cuantitativa
| Directa | Inversa | |
|---|---|---|
| ATF4 | 5′-TCTCCAGCGACAAGGCTAA-3’ | 5′-CCAATCTGTCCCGGAGAA-3’ |
| ATF6 | 5′-AATACTGAACTATGGACCTATGAGCA-3’ | 5′-TTGCAGGGCTCACACTAGG-3’ |
| ATG3 | 5′-CCTATTATGCCTAACTGGCACAT-3’ | 5′-TAGACAGTCTTCCAAGTTGCTACC-3’ |
| ATG4B | 5′-CAGATGATCTTTGCCCAAGC-3’ | 5′-TGGCTGCCTCTTCCTTTG-3’ |
| ATG14 | 5′-TGGGGACTACTCTGCCTACTACA-3’ | 5′-GGGTTACTCTGCTCCATGTCA-3’ |
| BCL-2 | 5′-AGCACGTGCACAGCTTCA-3’ | 5′-GTCCACGGGTGAAACAGC-3’ |
| proBNP | 5′-CGCAAAATGGTCCTCTACAC-3’ | 5′-CCGTGGAAATTTTGTGCTC-3’ |
| CASP3 | 5′-TGAGTGCTCGCAGCTCATA-3’ | 5′-GGGCTCGCTAACTCCTCAC-3’ |
| CHOP | 5′-TCACCACACCTGAAAGCAGA-3’ | 5′-TCTTGCAGGTCCTCATACCA-3’ |
| EDEM | 5′-CGGAAAGCGCTGGTAGAA-3’ | 5′-TGGAGTATTGTGGCTCGTCTT-3’ |
| GAPDH | 5′-AGCCACATCGCTCAGACAC-3’ | 5′-AATACGACCAAATCCGTTGACT-3’ |
| GRP78 | 5′-AATGACCAGAATCGCCTGAC-3’ | 5′-ATGCGCTCCTTGAGCTTTT-3’ |
| IL-1β | 5′GAAATCACACATGAACGTAGCC-3’ | 5′-CAAGTCATCCTCATTGCCACT-3’ |
| IRE1α | 5′-CCATCGAGCTGTGTGCAG-3’ | 5′-TGTTGAGGGAGTGGAGGTG-3’ |
| MAP1LC3B | 5′-GGTGTTGCCAGTTCCTGTCT-3’ | 5′-TCGCAAACCCAAGAGAAGTC-3’ |
| OS-9 | 5′-GGTCCAAGTGCGACCTTAAT-3’ | 5′-GTCCCCAGAGATACCTGCAC-3’ |
| PERK | 5′-TGGCCAGAAGTTGACAAAAGA-3’ | 5′-GGCAGCAATTCTCCCATCTA-3’ |
| P62 | 5′-ACATGGGGCTTGAGAAAGG-3’ | 5′-ATGCTACCGATGGTGAGGTC-3’ |
| TNNT2 | 5’-GGCTGCAGTGGCTACAGG-3’ | 5’CTGTCACCAGGCAATACAGC-3’ |
| ULK1 | 5′-CTGGCCGACTACCTGCAC-3’ | 5′-GGATGATGCCTTTGCTGTG-3’ |
| XBP1(s) | 5′-GCTGAGTCCGCAGCAGGT-3’ | 5′-GCTGAGTCCGCAGCAGGT-3’ |
La apoptosis se midió mediante la activación de la caspasa-3 y la caspasa-7 mediante el análisis FLICA (Immunochemistry Technologies, Estados Unidos) siguiendo las instrucciones del fabricante.
Determinación de viabilidad celularLa viabilidad celular se evaluó con el test de bromuro de 3-(4,5-dimetiltiazol-2-il)-2,5-difeniltetrazolio (MTT) (Sigma-Aldrich) según lo descrito con anterioridad18.
Determinación de autofagiaSe utilizó el kit de detección de autofagia CYTO-ID (Enzo Life Sciences, Estados Unidos) para medir la formación de autofagosomas siguiendo las instrucciones del fabricante.
Análisis de Western blot (inmunoelectrotransferencia)Se recolectaron las células AC16 transfectadas en tampón RIPA (Sigma-Aldrich), y se determinaron las concentraciones de proteína con un kit de análisis de proteína BCA (Thermo Fisher Scientific). Los anticuerpos primarios utilizados para el Western blot fueron los anti-caspasa-3, anti-caspasa-3-fragmentada, anti-GRP78, anti-LC3-B, anti-P62 (todos ellos de Cell Signaling Technology, Países Bajos), anti-ATG14 (Abcam, Reino Unido) y β-actina (Sigma-Aldrich). Las bandas proteicas se visualizaron con el sistema de detección de inmunotransferencia ECL (Bio-Rad, Estados Unidos) y se revelaron en un sistema de imagen ChemiDoc MP (Bio-Rad). Para analizar la expresión proteica, se cuantificaron las bandas mediante densitometría utilizando el programa informático de análisis Image Lab (Bio-Rad).
Análisis estadísticoLos datos se expresan en forma de media±error estándar de la media (EEM) salvo otra indicación. La significación estadística se definió como p <0,05. El análisis de las diferencias entre los individuos sanos, los pacientes con MCDi y los pacientes con MCD familiar se realizaron con el análisis de la varianza (ANOVA). Se utilizaron curvas de características operativas del receptor (ROC) para caracterizar el rendimiento diagnóstico del miR-16 mediante la determinación del área bajo la curva (AUC), la especificidad y la sensibilidad de los valores de corte óptimos. Las curvas ROC se generaron representando gráficamente la sensibilidad frente a 100 − especificidad. Los datos se presentan en forma de AUC e intervalo de confianza del 95% (IC95%). Se utilizó la prueba de la t de Student para analizar las diferencias tras la sobreexpresión del miR-16 en las células AC16. Las relaciones entre los miRNA y la fracción de eyección del VI se evaluaron mediante regresión logística. Se utilizó la prueba de Wilcoxon y se usaron combinaciones iterativas entre el miR-16, además de covariables ecocardiográficas y clínicas, para la creación de varios modelos. Los cambios en los valores de p de sus variables se evaluaron con la prueba de Wald y el cociente de verosimilitud. Se utilizó el programa estadístico R para todos los análisis. Las figuras se generaron con el programa informático GraphPad Prism 6.
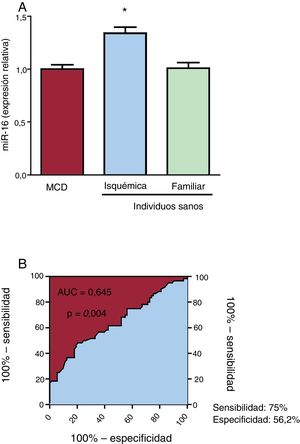
RESULTADOSEl miR-16 está aumentado en el plasma de los pacientes con miocardiopatía dilatada isquémicaLas características de los pacientes se muestran en la tabla 2. La expresión del miR-16 fue 1,34 veces mayor en el plasma de los pacientes con MCDi que en el de los controles sanos (p = 0,039) (figura 1A). Cabe señalar que no se observaron diferencias en las concentraciones plasmáticas de miR-16 entre los pacientes con MCD familiar y los individuos sanos. El análisis ROC del miR-16 mostró un AUC significativa (figura 1B). La potencia de discriminación para diferenciar a los pacientes con MCDi de los controles fue 0,645 (IC95%, 0,55-0,74; p=0,004). Con este valor de corte se obtenía una sensibilidad del 75,0% y una especificidad del 56,2% para la identificación de los pacientes con MCDi y los controles sanos.
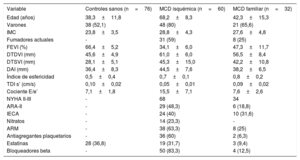
Variables demográficas, clínicas y ecocardiográficas de la población del estudio
| Variable | Controles sanos (n=76) | MCD isquémica (n=60) | MCD familiar (n=32) |
|---|---|---|---|
| Edad (años) | 38,3±11,8 | 68,2±8,3 | 42,3±15,3 |
| Varones | 38 (52,1) | 48 (80) | 21 (65,6) |
| IMC | 23,8±3,5 | 28,8±4,3 | 27,6±4,8 |
| Fumadores actuales | - | 31 (59) | 8 (25) |
| FEVI (%) | 66,4±5,2 | 34,1±6,0 | 47,3±11,7 |
| DTDVI (mm) | 45,6±4,9 | 61,0±6,0 | 56,5±8,4 |
| DTSVI (mm) | 28,1±5,1 | 45,3±15,0 | 42,2±10,8 |
| DAI (mm) | 36,4±8,3 | 44,5±7,6 | 38,2±6,5 |
| Índice de esfericidad | 0,5±0,4 | 0,7±0,1 | 0,8±0,2 |
| TDI s’ (cm/s) | 0,10±0,02 | 0,05±0,01 | 0,09±0,02 |
| Cociente E/e’ | 7,1±1,8 | 15,5±7,1 | 7,6±2,6 |
| NYHA II-III | - | 68 | 34 |
| ARA-II | - | 29 (48,3) | 6 (18,8) |
| IECA | - | 24 (40) | 10 (31,6) |
| Nitratos | - | 14 (23,3) | - |
| ARM | - | 38 (63,3) | 8 (25) |
| Antiagregantes plaquetarios | - | 36 (60) | 2 (6,3) |
| Estatinas | 28 (36,8) | 19 (31,7) | 3 (9,4) |
| Bloqueadores beta | - | 50 (83,3) | 4 (12,5) |
ARA-II: antagonistas del receptor de la angiotensina II; ARM: antagonistas del receptor de mineralocorticoides; DAI: dimensión de la aurícula izquierda; DTDVI: dimensión telediastólica del ventrículo izquierdo; DTSVI: dimensión telesistólica del ventrículo izquierdo; FEVI: fracción de eyección del ventrículo izquierdo; IECA: inhibidores de la enzima de conversión de la angiotensina; IMC: índice de masa corporal; MCD: miocardiopatía dilatada; NYHA: clase funcional de la New York Heart Association; TDI s’: onda sistólica máxima de la imagen de Doppler tisular.
Los valores expresan media ± error estándar de la media o n (%).
 en plasma detectada mediante reacción en cadena de la polimerasa en tiempo real cuantitativa (qRT-PCR) en individuos sanos y pacientes con miocardiopatía dilatada (MCD) isquémica o familiar. A: la expresión de miR-16 aumentó significativamente en los pacientes con MCD isquémica (n=60) en comparación con los pacientes con MCD familiar (n=32) y los individuos sanos (n=76). B: análisis de características operativas del receptor (ROC) del miR-16 para la MCD isquémica, con un área bajo la curva (AUC) = 0,645 (intervalo de confianza del 95%, 0,55-0,74; p=0,004), una sensibilidad del 75% y una especificidad del 56,2%.*p <0,05.")
Expresión del microRNA-16-5p (miR-16) en plasma detectada mediante reacción en cadena de la polimerasa en tiempo real cuantitativa (qRT-PCR) en individuos sanos y pacientes con miocardiopatía dilatada (MCD) isquémica o familiar. A: la expresión de miR-16 aumentó significativamente en los pacientes con MCD isquémica (n=60) en comparación con los pacientes con MCD familiar (n=32) y los individuos sanos (n=76). B: análisis de características operativas del receptor (ROC) del miR-16 para la MCD isquémica, con un área bajo la curva (AUC) = 0,645 (intervalo de confianza del 95%, 0,55-0,74; p=0,004), una sensibilidad del 75% y una especificidad del 56,2%.*p <0,05.
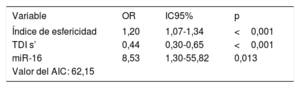
Los análisis de regresión logística multivariable mostraron que las concentraciones circulantes de miR-16 mostraban una asociación con el índice de esfericidad y con la onda sistólica máxima de la imagen de Doppler tisular (TDI s’). Según lo indicado por este análisis, el índice de esfericidad (odds ratio [OR] = 1,20; IC95%, 1,07-1,34; p < 0,001), TDI s’ (OR = 0,44; IC95%, 0,30-0,65; p <0,001) y la concentración plasmática de miR-16 (OR = 8,53; IC95%, 1,30-55,82; p=0,013) son factores independientes asociados con la MCDi (tabla 3). El criterio de información de Akaike mostró un valor de 62,15. El análisis ROC del modelo multivariante mostró un AUC = 0,963 (IC95%, 0,92-1,00; p <0,001).
Análisis de regresión logística multivariable de los pacientes con MCDi
| Variable | OR | IC95% | p |
|---|---|---|---|
| Índice de esfericidad | 1,20 | 1,07-1,34 | <0,001 |
| TDI s’ | 0,44 | 0,30-0,65 | <0,001 |
| miR-16 | 8,53 | 1,30-55,82 | 0,013 |
| Valor del AIC: 62,15 |
AIC: criterio de información de Akaike; IC95%: intervalo de confianza del 95%; MCDi: miocardiopatía dilatada isquémica; OR: odds ratio; TDI s’: onda sistólica máxima de la imagen de Doppler tisular.
El modelo incluía el índice de esfericidad, la TDI s’ y la concentración plasmática de miR-16.
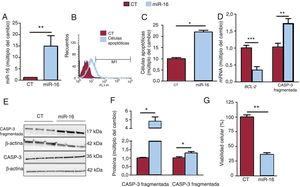
Las concentraciones de miR-16 aumentaron (figura 2A), y esta sobrexpresión indujo la apoptosis (cambio de 2,187 veces en las células apoptóticas; p=0,02) (figura 2B,C). Estos resultados concuerdan con las menores concentraciones de mRNA de BCL-2 (cambio de 0,35 veces; p=0,0007) (figura 2D) y las superiores concentraciones proteicas de la caspasa-3 fragmentada (cambio de 4,82 veces; p=0,02) (figura 2E,F). Además, el miR-16 redujo la viabilidad celular en las células AC16 con sobrexpresión de miR-16 (una reducción del 64%; p=0,008) (figura 2G).
 fomenta la apoptosis en las células cardiacas humanas. A: se detectó la expresión relativa de mRNA de miR-16 mediante reacción en cadena de la polimerasa en tiempo real cuantitativa (qRT-PCR) y se normalizó respecto a la de SnRNA U6 (n=5). B: se determinó la presencia de células apoptóticas mediante el análisis FLICA con citometría de flujo. C: se muestran los resultados de la citometría de flujo cuantitativa de células cardiacas apoptóticas (n=3). D: se detectaron las concentraciones relativas de mRNA de BCL-2 y de mRNA de caspasa-3 mediante qRT-PCR y se normalizaron respecto a los de GAPDH (n=7). E: se sometieron lisados de células enteras a un análisis de inmunotransferencia con anticuerpos específicos contra la caspasa-3 fragmentada, la caspasa-3 y la β-actina (n=3). F: cuantificación de la caspasa-3 fragmentada y la caspasa-3 normalizada respecto a la β-actina (n=3). G: se evaluó la viabilidad celular mediante un análisis MTT; los valores se presentan como porcentaje de viabilidad celular. CASP-3: caspasa-3; CT: células AC16 de control; miR-16: células AC16 con sobrexpresión de microRNA-16-5p. *p < 0,05. **p < 0,01. ***p < 0,005.")
La sobrexpresión del microRNA-16-5p (miR-16) fomenta la apoptosis en las células cardiacas humanas. A: se detectó la expresión relativa de mRNA de miR-16 mediante reacción en cadena de la polimerasa en tiempo real cuantitativa (qRT-PCR) y se normalizó respecto a la de SnRNA U6 (n=5). B: se determinó la presencia de células apoptóticas mediante el análisis FLICA con citometría de flujo. C: se muestran los resultados de la citometría de flujo cuantitativa de células cardiacas apoptóticas (n=3). D: se detectaron las concentraciones relativas de mRNA de BCL-2 y de mRNA de caspasa-3 mediante qRT-PCR y se normalizaron respecto a los de GAPDH (n=7). E: se sometieron lisados de células enteras a un análisis de inmunotransferencia con anticuerpos específicos contra la caspasa-3 fragmentada, la caspasa-3 y la β-actina (n=3). F: cuantificación de la caspasa-3 fragmentada y la caspasa-3 normalizada respecto a la β-actina (n=3). G: se evaluó la viabilidad celular mediante un análisis MTT; los valores se presentan como porcentaje de viabilidad celular. CASP-3: caspasa-3; CT: células AC16 de control; miR-16: células AC16 con sobrexpresión de microRNA-16-5p. *p < 0,05. **p < 0,01. ***p < 0,005.
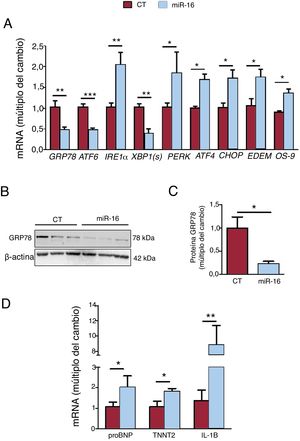
A continuación se analizaron la cantidad de los genes clave involucrados en el estrés del RE, la respuesta de degradación de proteínas relacionadas con el RE (ERAD) y los biomarcadores de IC (figura 3A-C). La sobrexpresión de miR-16 fomentó la cascada del estrés del RE apoptótico, dado el grado de expresión de mRNA de la vía de PERK/CHOP: PERK (cambio de 1,84 veces; p=0,01), ATF4 (cambio de 1,68 veces; p=0,02) y CHOP (cambio de 1,72 veces; p=0,03) (figura 3A). En cambio, se observó una disminución de GRP78 (mRNA, cambio de 0,47 veces; p=0,004; proteína, cambio de 0,22 veces; p=0,04) y mRNA de ATF6 (cambio de 0,48 veces; p=0,002) y XBP1(s) (cambio de 0,38 veces; p=0,01). Además, la sobrexpresión de miR-16 activó la respuesta de ERAD (mRNA de EDEM, cambio de 1,74 veces; p=0,03; mRNA de OS-9, cambio de 1,36 veces; p=0,02). Por lo que respecta a los biomarcadores de IC, la sobrexpresión de miR-16 fomentó la expresión del propéptido natriurético cerebral (proBNP) (cambio de 2,03 veces; p=0,03) y la troponina T miocárdico (TNNT2) (cambio de 1,83 veces; p=0,02) (figura 3C). Además, la concentración de mRNA de IL-1β fue 8,84 veces superior (p=0,035) (figura 3C).
 en el estrés del retículo endoplásmico (RE), los biomarcadores de insuficiencia cardiaca y la inflamación en células cardiacas humanas. A: se detectaron los valores relativos de mRNA de los marcadores del estrés del RE (GRP78, ATF6, IRE1α, XBP1(s), ATF4, CHOP, EDEM y OS-9) mediante reacción en cadena de la polimerasa en tiempo real cuantitativa (qRT-PCR) y se normalizaron respecto a los de GAPDH (n=5). B: se sometieron lisados de células enteras a un análisis de inmunotransferencia con anticuerpos específicos contra GRP78 y β-actina (n=3). C: cuantificación de las concentraciones de proteína GRP78, normalizadas respecto a las de β-actina (n=3). D: se detectaron concentraciones relativas de biomarcadores de la IC (propéptido natriurético cerebral y TNNT2) y de inflamación (IL-1B) mediante qRT-PCR y se normalizaron respecto a los de GAPDH (n=5). CT: células AC16 de control; miR-16: células AC16 con sobrexpresión de microRNA-16-5p. *p < 0,05. **p < 0,005.")
Efectos del microRNA-16-5p (miR-16) en el estrés del retículo endoplásmico (RE), los biomarcadores de insuficiencia cardiaca y la inflamación en células cardiacas humanas. A: se detectaron los valores relativos de mRNA de los marcadores del estrés del RE (GRP78, ATF6, IRE1α, XBP1(s), ATF4, CHOP, EDEM y OS-9) mediante reacción en cadena de la polimerasa en tiempo real cuantitativa (qRT-PCR) y se normalizaron respecto a los de GAPDH (n=5). B: se sometieron lisados de células enteras a un análisis de inmunotransferencia con anticuerpos específicos contra GRP78 y β-actina (n=3). C: cuantificación de las concentraciones de proteína GRP78, normalizadas respecto a las de β-actina (n=3). D: se detectaron concentraciones relativas de biomarcadores de la IC (propéptido natriurético cerebral y TNNT2) y de inflamación (IL-1B) mediante qRT-PCR y se normalizaron respecto a los de GAPDH (n=5). CT: células AC16 de control; miR-16: células AC16 con sobrexpresión de microRNA-16-5p. *p < 0,05. **p < 0,005.
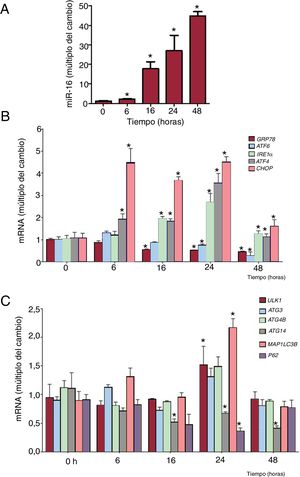
Se realizaron experimentos de evolución temporal para caracterizar mejor la activación del estrés del RE inducido por el miR-16 (figura 4). La cantidad de mRNA de IRE1α (cambio de 1,84 veces, p=0,02), ATF4 (cambio de 1,96 veces; p=0,03) y CHOP (cambio de 3,68 veces; p=0,002) aumentó significativamente a las 6-16 h de la transfección de miR-16. La concentración de mRNA de ATF6 y de GRP78 disminuyó en respuesta a la sobrexpresión de miR-16 (figura 4A,B). A continuación se analizaron los transcriptos relacionados con la autofagia. Los mRNA de ULK1 (cambio de 1,57 veces; p=0,04) y MAP1LC3B (cambio de 2,17 veces; p=0,04) habían aumentado significativamente 24 h después de la transfección de miR-16. Los mRNA de ATG14 se redujeron en sobrexpresión de miR-16.
 fomenta el estrés del retículo endoplásmico (RE) antes de la autofagia de las células cardiacas humanas. A: se detectaron los valores relativos de miR-16 mediante reacción en cadena de la polimerasa en tiempo real cuantitativa (qRT-PCR) y se normalizó respecto a los de SnRNA U6 (n=4). B: se detectaron los valores relativos de mRNA de los marcadores del estrés del RE (GRP78, ATF6, IRE1α, ATF4 y CHOP) mediante qRT-PCR y se normalizaron respecto a los de GAPDH (n=4). C: se detectaron los valores relativos de mRNA de los marcadores de la autofagia (ULK1, ATG3, ATG4B, ATG14, MAP1LC3B y p62) mediante qRT-PCR y se normalizaron respecto a los de GAPDH (n=4).*p <0,05.")
La sobrexpresión de microRNA-16-5p (miR-16) fomenta el estrés del retículo endoplásmico (RE) antes de la autofagia de las células cardiacas humanas. A: se detectaron los valores relativos de miR-16 mediante reacción en cadena de la polimerasa en tiempo real cuantitativa (qRT-PCR) y se normalizó respecto a los de SnRNA U6 (n=4). B: se detectaron los valores relativos de mRNA de los marcadores del estrés del RE (GRP78, ATF6, IRE1α, ATF4 y CHOP) mediante qRT-PCR y se normalizaron respecto a los de GAPDH (n=4). C: se detectaron los valores relativos de mRNA de los marcadores de la autofagia (ULK1, ATG3, ATG4B, ATG14, MAP1LC3B y p62) mediante qRT-PCR y se normalizaron respecto a los de GAPDH (n=4).*p <0,05.
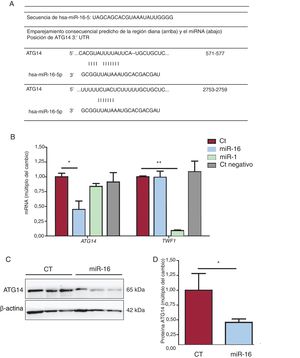
Los instrumentos de predicción bioinformáticos (TargetScan, miRBase y miRDB)19 revelaron la secuencia de unión de la interacción miRNA-mRNA (figura 5A). El grado de expresión de mRNA de ATG14 y de TWF1 se redujo en 0,448 y 0,091 veces, respectivamente, tras la sobrexpresión de miR-16 y miR-1 (figura 5B). Estos resultados se corroboraron con la reducción de las concentraciones de proteína ATG14 en comparación con lo observado en los controles (cambio de 0,16 veces; p=0,03) (figura 6C), lo cual confirmó nuevamente que el miR-16 puede producir una regulación negativa de la expresión de ATG14 en las células AC16.
 y se normalizaron respecto a los de GAPDH (n=4). C: se sometieron lisados de células enteras a un análisis de inmunotransferencia con anticuerpos específicos contra ATG14 and β-actina. D: cuantificación de las concentraciones de ATG14, normalizadas respecto a las de β-actina (n=3). CT: células AC16 de control; miR-1: células AC16 con sobrexpresión del control positivo de microRNA-1; miR-16: células AC16 con sobrexpresión de microRNA-16-5p; Ct negativo: células AC16 de control negativo. *p < 0,05.**p < 0,005.")
Análisis de la expresión génica de ATG14. A: emparejamiento de regiones diana de Atg14 y miR-16 predichas mediante TargetScan, miRBase y miRDB. B: se detectaron los valores relativos de mRNA de ATG14 y TWF1 mediante reacción en cadena de la polimerasa en tiempo real cuantitativa (qRT-PCR) y se normalizaron respecto a los de GAPDH (n=4). C: se sometieron lisados de células enteras a un análisis de inmunotransferencia con anticuerpos específicos contra ATG14 and β-actina. D: cuantificación de las concentraciones de ATG14, normalizadas respecto a las de β-actina (n=3). CT: células AC16 de control; miR-1: células AC16 con sobrexpresión del control positivo de microRNA-1; miR-16: células AC16 con sobrexpresión de microRNA-16-5p; Ct negativo: células AC16 de control negativo. *p < 0,05.**p < 0,005.
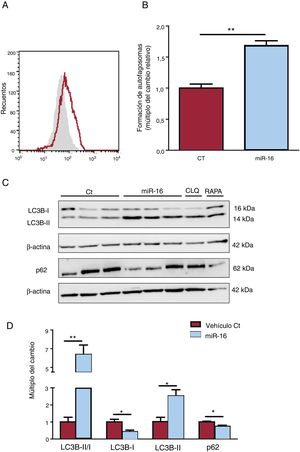
 en la autofagia de los cardiocitos humanos. A: se determinó la presencia de autofagosomas mediante CYTO-ID y citometría de flujo. B: se muestran los resultados de la citometría de flujo cuantitativa (n=3). C: se sometieron lisados de células enteras a un análisis de inmunotransferencia con anticuerpos específicos contra LC3B (I y II), p62 y β-actina (n=3). D: cuantificación del cociente LC3BII/I y de las concentraciones de p62, normalizadas ambas respecto a las de β-actina (n=3). CT: células AC16 de control; miR-16: células AC16 con sobrexpresión de microRNA-16-5p.*p < 0,05.**p < 0,005.")
Efectos del microRNA-16-5p (miR-16) en la autofagia de los cardiocitos humanos. A: se determinó la presencia de autofagosomas mediante CYTO-ID y citometría de flujo. B: se muestran los resultados de la citometría de flujo cuantitativa (n=3). C: se sometieron lisados de células enteras a un análisis de inmunotransferencia con anticuerpos específicos contra LC3B (I y II), p62 y β-actina (n=3). D: cuantificación del cociente LC3BII/I y de las concentraciones de p62, normalizadas ambas respecto a las de β-actina (n=3). CT: células AC16 de control; miR-16: células AC16 con sobrexpresión de microRNA-16-5p.*p < 0,05.**p < 0,005.
La sobrexpresión de miR-16 produce una acumulación de autofagosomas, tal como indica el aumento de la intensidad de tinción de CYTO-ID en comparación con los controles (cambio de 1,69 veces; p <0,001) (figura 6A,B). Se determinó el cociente LC3BI/II y la p62 como proteínas representativas del flujo autofágico (figura 6C,D). Se observó un aumento significativo de la conversión de LC3B-I a LC3B-II (cambio de 6,37 veces; p=0,003) y de la degradación de la proteína p62 (cambio de 0,74 veces; p=0,01) que reflejaba la actividad autofágica en las células cardiacas con sobrexpresión de miR-16.
DISCUSIÓNLa MCD es la miocardiopatía más frecuente y el principal motivo de los trasplantes de corazón en todo el mundo20. Se trata de una enfermedad compleja, con un fenotipo común que engloba mecanismos etiológicos heterogéneos. Las mejoras en la fisiopatología subyacente en la MCD podrían reducir la carga clínica de esta entidad. Así pues, continúa habiendo necesidad de nuevos indicadores no invasivos, como los miRNA, para facilitar la toma de decisiones clínicas y la elaboración de estrategias terapéuticas. Además, la identificación de miRNA anómalos y sus dianas podría revelar la patogenia subyacente a la MCDi a escala molecular3,21–26. Se observó que la expresión de miR-16 era significativamente mayor en el plasma de los pacientes con MCDi que en los individuos sanos o los pacientes con MCD familiar, lo cual confirma que el miR-16 está relacionado con la MCDi. Por otra parte, varios parámetros ecocardiográficos como el índice de esfericidad y la TDI s’ sistólica máxima mostraron una asociación con la cohorte de MCDi. El valor inferior del índice de esfericidad y la TDI s’ se han relacionado con un peor pronóstico en la MCD. Considerando los resultados conjuntamente con los obtenidos por Devaux et al.10, nosotros proponemos un modelo multiparamétrico que incluye el índice de esfericidad, la TDI s’ y el miR-16, que podría mejorar la identificación de la MCDi. En consecuencia, el miR-16 parece prometedor como biomarcador de la MCDi para una posible traslación al ámbito clínico.
La sobreexpresión de miR-16 inhibe la proliferación celular y fomenta la apoptosis mediante la acción directa sobre la BCL-2 en modelos de isquemia de rata11,12. En nuestro estudio, esta situación se simuló en una línea de cardiocitos humanos por lo que respecta a la apoptosis, la viabilidad celular y la BCL-2. En cambio, se ha descrito que la BCL-2 está aumentada y puede intervenir en un mecanismo de compensación en el VI de la población con MCD24. Por otra parte, se observa una activación de la muerte celular en el VI de los pacientes con una MCD en fase terminal27. Liu et al.27 señalaron que la caspasa-3 y la CHOP, que son proteínas clave involucradas en la muerte celular, estaban activadas en los tejidos del VI de pacientes con MCD en fase terminal, en comparación con lo observado en el tejido cardiaco normal. Nuestros datos demostraron que la sobreexpresión de miR-16 fomenta el aumento del mRNA de CHOP y de las concentraciones proteicas de caspasa-3 y caspasa-3 fragmentada. No obstante, será necesario disponer de más evidencia para determinar los eventos patológicos sucesivos que convergen en la apoptosis de los miocardiocitos.
La BCL-2, que es una de las proteínas antiapoptóticas esenciales, desempeña un papel central en la adaptación al estrés celular, incluida la interacción molecular entre el estrés del RE y la autofagia28,29. Dado que la BCL-2 es un objetivo principal del miR-16, se analizaron estas dos respuestas de adaptación celular. Diversas observaciones indican que la secuencia proapoptótica relacionada con el estrés del RE se ve influida por el miR-16 en las células cardiacas humanas. En primer lugar, la sobrexpresión de las concentraciones de CHOP conduce a apoptosis celular y detención del ciclo celular30,31. Nuestros resultados confirmaron que la CHOP aumenta poco después de la sobrexpresión de miR-16, con un incremento posterior en el número de células cardiacas apoptóticas. La CHOP regula varios genes proapoptóticos y antiapoptóticos, incluida la transcripción de la caspasa-3 y la BCL-232. En condiciones de estrés del RE, la expresión de la BCL-2 se reduce por la CHOP, que sensibiliza las células para la apoptosis33. En segundo lugar, la GRP78, que es un factor regulador principal de la citoprotección y reduce el estrés del RE, está aumentada tras una simulación isquémica en el modelo de miocardiocitos de rata34. Sin embargo, la sobrexpresión de miR-16 en nuestro modelo celular redujo las concentraciones de GRP78. Es posible que esto se deba a una regulación negativa del ATF6, gen diana del miR-1635 o a la inhibición general de la traslación proteica por la vía de PERK/CHOP.
Un estrés del RE abrumador fomenta la inflamación y la IC7,36. El proBNP, la forma prohormonal de los péptidos natriuréticos (BNP y NT-proBNP), y la TNNT2 son biomarcadores clínicos importantes para el diagnóstico de la disfunción cardiaca después de un evento isquémico37,38. Nosotros hemos confirmado que el miR-16 conduce a la expresión de proBNP yTNNT2. En situaciones patológicas (es decir, estrés mecánico o isquemia), las células del VI expresan el proBNP como respuesta a la expansión de volumen del ventrículo37. La TNNT2 es un elemento del aparato contráctil de los miocardiocitos. Regula la contracción muscular en respuesta a los cambios del calcio intracelular e indica el daño del músculo cardiaco de manera muy específica38. Por lo que respecta a la inflamación, las citocinas proinflamatorias como la IL-1β aumentan la permeabilidad de la membrana celular de los miocardiocitos y fomentan la apoptosis y la liberación de troponinas39. Miyazaki et al.40 demostraron que la vía que actúa a través de PERK/CHOP aumenta la inflamación tisular, incluida la IL-1β, tras la isquemia/reperfusión del miocardio en un modelo de ratón. La sobrexpresión de miR-16 conduce a un aumento del mRNA de la IL-1β. Además, la vía de PERK/CHOP desempeña un papel específico en el control de la producción de la IL-1β y de su maduración41. Por otra parte, las concentraciones de IL-1β permiten predecir la mortalidad y la necesidad de trasplante de corazón en los pacientes con MCD42.
El estrés del RE y la inflamación podrían iniciar el proceso de autofagia en la patogenia de las cardiopatías7,43. Nuestros datos demuestran que la autofagia está activada después del estrés del RE, posiblemente como respuesta a proteínas con un plegamiento alterado o con agregación como consecuencia de la sobrexpresión de miR-16. La presencia de autofagosomas en la población con MCD idiopática indica una mejora del pronóstico de la IC. Estos resultados indican una función protectora de la autofagia en cuanto al deterioro del miocardio8,9. Hemos confirmado que el miR-16 fomenta la acumulación de autofagosomas. Hay más de 30 proteínas de genes relacionados con la autofagia (ATG) que intervienen en ella, en especial en la formación de autofagosomas29, y la ATG14 es una de las principales proteínas involucradas44. Además, el ATG14 es uno de los principales genes diana regulados por el miR-16 en las células del endotelio vascular45. En el presente estudio, el miR-16 inhibió la progresión de la autofagia y participó en la lesión del endotelio vascular y la cardiopatía crónica45. En nuestro modelo, hubo una significativa regulación negativa de la ATG14 tras la sobrexpresión del miR-16. Sin embargo, el flujo autofágico se activó, y ello indica que es posible que la ATG14 no tuviera un papel crucial en este proceso. Recientemente, Caragnano et al.46 han afirmado que el proceso de la autofagia está bloqueado en los pacientes con MCD, dada la acumulación de proteína p62. Así pues, el papel de la autofagia como programa de citoprotección o tendente a la muerte celular continúa sin haberse determinado en las diversas etiologías de la MCD. Nosotros proponemos que el miR-16 fomenta la autofagia celular y reduce la agregación de proteínas como arma final para afrontar el problema. De hecho, se ha descrito que la isquemia crónica activa la autofagia47 y que su estimulación protege a los cardiomiocitos del daño isquémico48. Sin embargo, la regulación positiva abrumadora de la autofagia podría conducir a apoptosis de los miocardiocitos y un empeoramiento de la función cardiaca43.
En resumen, nuestros resultados confirman que el miR-16 hace un papel importante en la progresión del daño de las células cardiacas humanas en la MCDi. Al actuar a través de varias dianas moleculares, el miR-16 tiene una repercusión secuencial en el estrés del RE, la inflamación, la autofagia y la apoptosis. Así pues, una mejora del conocimiento sobre la interacción entre estos procesos y el esclarecimiento del papel del miR-16 facilitarán el desarrollo de estrategias terapéuticas alternativas e individualizadas para la MCDi.
LimitacionesEl estudio tiene ciertas limitaciones. En primer lugar, la utilidad de la miR-16 deberá compararse con la de las pruebas bioquímicas actualmente establecidas. En segundo lugar, dada la ausencia de muestras de biopsia cardiaca, fue imposible determinar si las altas concentraciones plasmáticas de miR-16 reflejaban el patrón de expresión de miRNA intracelular. Aunque el uso de una línea de cardiocitos humanos estable tiene ciertas ventajas y es esencial para describir los procesos celulares y moleculares relacionados con la sobreexpresión de miR-16, serán necesarias nuevas investigaciones para la traslación clínica de estos resultados. Aunque hubo una regulación negativa de la expresión de ATG14 después de la sobrexpresión de miR-16, deberá realizarse un análisis de luciferasa para determinar si la ATG14 se ve afectada directamente por el miR-16 en esta línea de cardiocitos humanos.
CONCLUSIONESLas concentraciones plasmáticas del miR-16 muestran un aumento en los pacientes con MCDi, lo cual confirma su relación con esta etiología. Nuestro estudio revela que el miR-16 activa el estrés del RE y fomenta la inflamación seguida de autofagia de las células cardiacas humanas en condiciones de isquemia. Así pues, la autofagia puede ser un intento de mantener la homeostasis celular en respuesta a proteínas mal plegadas o agregadas en relación con el estrés del RE previo a la apoptosis.
FINANCIACIÓNEste trabajo fue financiado por subvenciones concedidas en el marco de la Iniciativa Territorial Integrada (ITI PI0048-2017 e ITI0033_2019), la FPS00136-2018 y una subvención de investigación clínica de la Sociedad Española de Cardiología para la Investigación Básica en Cardiología (PI0012_2019).
CONFLICTO DE INTERESESR. Toro y A. Mangas han presentado una solicitud de patente sobre el uso de microRNA como biomarcadores. Los demás autores no tienen nada que declarar.
- –
Pese a ser una de las principales causas de la IC, la fisiopatología de la MCDi a escala subcelular continúa sin estar clara.
- –
Cada vez hay más evidencia de que hay una desregulación de miRNA en varias enfermedades cardiovasculares, incluida la MCD.
- –
La concentración de miR-16 está aumentada en modelos de la MCDi de rata y en pacientes con MCDi.
- –
Su sobrexpresión desencadena la apoptosis miocárdica a través de una regulación postranscripcional de la BCL-2 en un modelo de isquemia miocardiocitaria de rata.
- –
Además, su ausencia en los miocardiocitos mejora la función cardiaca.
- –
Las concentraciones circulantes de miR-16 fueron significativamente mayores en el plasma de los pacientes con MCDi que en los controles sanos y no mostraron ningún aumento en los pacientes con MCD familiar, lo cual indica que el miR-16 está relacionado específicamente con la MCDi.
- –
Además, nuestros resultados indican que el miR-16 induce el estrés del RE, marcadores de la IC, inflamación y autofagia y en las células cardiacas humanas.
- –
Una mejora del conocimiento sobre estos procesos y el esclarecimiento del papel del miR-16 facilitarán el desarrollo de estrategias terapéuticas para la MCDi.
Damos las gracias a todo el personal clínico que participó en el estudio y a Lucille Banham por su valiosa ayuda en la preparación del manuscrito en inglés.