
A menudo, los aneurismas de la aorta torácica son asintomáticos y permanecen sin diagnosticar hasta que surgen complicaciones catastróficas, como la disección aórtica, que conllevan una muy elevada mortalidad1.
Actualmente se conoce que ciertas variantes genéticas predisponen a enfermedad de la raíz aórtica y la aorta ascendente, en los denominados aneurismas y disecciones de la aorta torácica hereditarios (ADATH). Así, el estudio genético (EG) y el hallazgo de estas variantes pueden ser de gran utilidad para identificar a los individuos en riesgo2.
Los ADATH clásicamente se han considerado sindrómicos (p. ej., los síndromes de Marfan, de Loeys-Dietz o de Ehlers-Danlos tipo vascular) o no sindrómicos (el aneurisma de la aorta torácica es un hallazgo aislado). Sin embargo, la distinción entre ambos se ha difuminado al demostrarse una muy amplia variabilidad fenotípica en muchas de las variantes descubiertas.
Postulamos que disponer de una consulta específica de aortopatías hereditarias (AH) favorece la correcta identificación de estos pacientes, implementando de una forma más completa y eficiente el EG.
En este análisis retrospectivo unicéntrico se incluyó a los pacientes estudiados en la consulta de AH de un hospital de tercer nivel entre 2010 y 2020. En una primera visita se efectuó una minuciosa exploración física para hallar signos clínicos clásicamente asociados con ADATH sindrómicos y un ecocardiograma transtorácico para obtener las medidas de la raíz aórtica y la aorta ascendente según las recomendaciones vigentes3. Tras ello, se extrajo una muestra sanguínea o de saliva para EG a los pacientes con: diámetros aórticos ajustados al tamaño corporal y la edad superiores a la normalidad (Z-score> 2) en ausencia de otros factores de riesgo; diámetros aumentados con otros rasgos fenotípicos clásicamente asociados con ADATH sindrómicas o antecedentes familiares de eventos aórticos o muerte súbita; o familiares de primer grado de pacientes con alguna de las características anteriores y/o de los que ya se conocía una variante genética posiblemente asociada con aortopatía. Los EG se realizaron combinando los métodos de secuenciación masiva (NGS) y Sanger. Se denominó caso índice al primer paciente de una familia que acudió a la consulta. En el caso de hallarse una variante genética con cierta probabilidad de ser patogénica, se inició el cribado «en cascada», ofreciendo el EG a todos los familiares de primer grado. Únicamente no se realizó en el caso de pacientes que se negaron o cuya relación de primer grado no estaba asegurada. El estudio fue aprobado por el Comité de Ética local y los pacientes firmaron un consentimiento informado por escrito antes de la extracción de la muestra para EG.
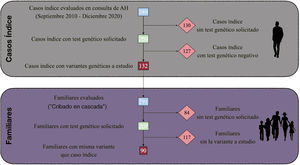
Tras evaluarse a 389 pacientes índice, se solicitó EG dirigido a 259 (66,6%). De ellos, en 132 (50,9%) se obtuvo una variante genética de interés (figura 1), siendo 46 patogénicas o probablemente patogénicas (17,7%). Estos hallazgos llevaron a evaluar a 291 familiares, de los que a 207 (71,1%) se les solicitó EG; 90 (43,5%) obtuvieron un resultado positivo para la variante estudiada. Así, se descartó la presencia de dicha variante en 117 familiares. A los pacientes con variantes halladas, patogénicas o no, así como aquellos con EG negativo pero con persistencia de sospecha de predisposición a aortopatía, se les realizó un seguimiento periódico.

Finalmente, 140 pacientes portaban variantes en FBN1, 11 en genes codificantes de TGF-B o sus receptores, 19 en los de COL3A1, 22 en los de otras proteínas formadoras de colágeno y 22 en genes codificantes de proteínas de las células de músculo liso vascular. Sus características, así como las características clínicas de los pacientes portadores, se presentan en la tabla 1.
Características de los pacientes con sospecha de aneurismas y disecciones de la aorta torácica hereditarios y hallazgo de una variante de interés en el estudio genético
| Total (N=222) | Variantes en FBN1 (n=140) | Variantes en TGFBR1, TGFBR2 o TGB3 (n=11) | Variantes en Col3A1 (n=19) | Variantes en otros genes codificantes de colágenos (1A1, 1A2, 5A1) (n=22) | Variantes en otros genes codificantes de proteínas de la CMLV (ACTA, JAG…) (n=22) | |
|---|---|---|---|---|---|---|
| Criterios diagnósticos de síndrome | ||||||
| Síndrome de Marfan | 125 (56,3) | 125 (89,3) | — | — | — | — |
| Síndrome de Loeys-Dietz | 10 (4,5) | — | 10 (90,9) | — | — | — |
| Síndrome de Ehlers-Danlos tipo vascular | 14 (0,6) | — | — | 14 (73,7) | — | — |
| Casos índice | 132 (59,5) | 86 (61,4) | 6 (54,5) | 8 (42,1) | 12 (54,5) | 12 (54,5) |
| Familiares portadores | 90 (40,5) | 54 (38,6) | 5 (45,5) | 11 (57,9) | 10 (45,5) | 10 (45,5) |
| Edad (años) | 34,8±18,1 | 32,7±17,4 | 31,0±19,1 | 39,1±18,9 | 34,1±18,1 | 45,2±14,9 |
| Varones | 118 (53,2) | 72 (51,4) | 7 (63,6) | 8 (42,1) | 13 (59,1) | 15 (68,2) |
| Aorta ascendente | ||||||
| Dilatación aórtica leve | 105 (47,3) | 79 (56,4) | 1 (9,1) | 7 (36,8) | 7 (31,8) | 6 (27,3) |
| Aneurisma de la aorta torácica ascendente | 39 (17,6) | 26 (18,6) | 5 (45,5) | 0 | 2 (9,1) | 5 (22,7) |
| Disección tipo A | 13 (5,9) | 11 (7,9) | 0 | 0 | 0 | 2 (9,1) |
| Rotura aórtica | 1 (0,5) | 0 | 1 (5,3) | 0 | 0 | 0 |
| Intervención | 48 (21,6) | 34 (24,3) | 4 (36,4) | 0 | 2 (9,1) | 7 (31,8) |
| Bono-Bental | 20 (41,7) | 14 (41,5) | 1 (25) | 0 | 0 | 5 (71,4) |
| Yacoub | 15 (31,3) | 11 (32,4) | 2 (50) | 0 | 0 | 2 (9,1) |
| David | 11 (22,9) | 9 (26,5) | 1 (25) | 0 | 0 | 1 (14,3) |
| Aneurismas otras localizaciones | 7 (3,2) | 0 | 2 (10,6) | 2 (10,5) | 2 (9,1) | 0 |
| Válvula aórtica bicúspide | 4 (1,8) | 2 (1,4) | 0 | 0 | 0 | 1 (4,5) |
| Prolapso de válvula mitral | 59 (26,6) | 48 (34,3) | 1 (9,1) | 7 (36,8) | 2 (9,1) | 0 |
| Ectopia lentis | 46 (320,7) | 46 (32,9) | 0 | 0 | 0 | 0 |
| Características de variantes genéticas | ||||||
| Patogénica/probablemente patogénica* | 146 (65,7) | 121 (86,4) | 9 (81,8) | 12 (63,2) | 0 | 1 (4,5) |
| Posiblemente patogénica | 6 (2,7) | 1 (0,7) | 1 (9,1) | 0 | 0 | 4 (18,2) |
| VUS | 70 (31,5) | 18 (12,9) | 1 (9,1) | 7 (36,8) | 22 (100) | 17 (77,3) |
| Previamente descrita en la literatura | 76 (34,2) | 66 (47,1) | 4 (36,4) | 1 (5,3) | 0 | 4 (18,2) |
Las variantes genéticas identificadas se nombran siguiendo las recomendaciones de la Human Genome Variation Society (HGVS) y el American College of Medical Genetics (ACMG). En el caso específico de las variantes de FBN1, estas se clasificaron como patogénicas si cumplían los criterios para mutaciones causales de FBN1 según los criterios modificados de Gante.
Las variables cualitativas se expresan como n (%) y las cuantitativas, como la media±desviación estándar.
Por lo tanto, más del 50% de las solicitudes obtuvieron una variante de interés. Renner et al.4 comunicaron un 40,7% en 199 individuos con características clínicas de ADATH sindrómicos o no sindrómicos. En cambio, las series de pacientes no seleccionados, con el único antecedente de aneurisma o disección de aorta torácica, reportan un 3,9-5% de positividad para variantes patogénicas o probablemente patogénicas y alrededor del 25% si incluyen variantes de significado incierto (VUS)5. Ello demuestra la importancia de guiar clínicamente la solicitud del EG. Además, en nuestro estudio, se reconoció a 90 familiares portadores de las variantes en estudio, lo que podría tener implicaciones directas en su seguimiento, tratamiento médico, reparación quirúrgica profiláctica y plan reproductivo.
Cabe destacar que el mayor porcentaje de variantes patogénicas o probablemente patogénicas se obtuvo en FBN1, gen asociado al síndrome de Marfan, la AH mejor estudiada. Resulta de interés un seguimiento de los pacientes con VUS que incremente el conocimiento acerca del posible significado pronóstico de estas variantes. Iniciativas como la Red Española de Patología Aórtica Genética (REPAG) sin duda ayudarán a este fin.
En definitiva, la existencia de una consulta específica de AH puede ayudar a guiar un tratamiento individualizado de estos pacientes, así como a dar sentido a la ingente cantidad de información obtenida de los EG.
FINANCIACIÓNNo se recibió financiación para la elaboración de este estudio.
CONTRIBUCIÓN DE LOS AUTORESV.M. Becerra-Muñoz realizó los análisis y redactó el manuscrito; V.M. Becerra-Muñoz, A. Díaz-Expósito y V. Doncel-Abad recogieron los datos clínicos y genéticos; P. Fernández-García, J.L. López-Benítez y F. Cabrera-Bueno atendieron a los pacientes que visitaron la consulta de aortopatías hereditarias; F. Cabrera-Bueno es el coordinador de la unidad de aortopatías hereditarias; V.M. Becerra-Muñoz, A. Díaz-Expósito, V. Doncel-Abad, P. Fernández-García, J.L. López-Benítez y F. Cabrera-Bueno revisaron y corrigieron el contenido final del manuscrito.
CONFLICTO DE INTERESESLos autores declaran que no presentan ningún conflicto de intereses en relación con este trabajo.