
Desde hace siglos sabemos que muchas características de los seres vivos se heredan. En los últimos años se ha avanzado mucho en el conocimiento de muchas enfermedades mendelianas causadas por mutaciones en uno o en un reducido número de genes. Sin embargo, el conocimiento de las bases genéticas de las enfermedades complejas, entre ellas la cardiopatía isquémica, está siendo mucho más dificultoso. En esta revisión de carácter narrativo, tenemos como objetivo revisar algunos de los resultados más relevantes obtenidos hasta la fecha sobre el estudio del componente genético de la cardiopatía isquémica. Presentamos los resultados más importantes que han aportado los estudios de ligamiento y asociación, incluidos los estudios de asociación integral del genoma. Por último, revisamos la utilidad que estos resultados pueden tener en la práctica clínica diaria y la prevención, y los retos previsibles para el futuro inmediato.
Palabras clave
Desde hace siglos sabemos que muchas características de los seres vivos se heredan. La genética es la parte de la ciencia que estudia la herencia y lo relacionado con ella. Aunque en 1866 Mendel ya describió las principales leyes de la genética, no fue posible avanzar en el estudio de las bases genéticas de las distintas enfermedades hasta que se desarrollaron métodos para secuenciar el ácido desoxirribonucleico (ADN)1–3. Estas y otras técnicas han permitido que en los últimos años hayamos avanzado hasta la secuenciación completa del genoma humano4, el conocimiento del patrón de desequilibrio de ligamiento en el genoma5 y en el genotipado a gran escala. Todos estos métodos y hallazgos han permitido avanzar en el conocimiento de las bases genéticas de diferentes enfermedades.
Actualmente, se han establecido las bases genéticas de más de 2.000 enfermedades monogénicas y oligogénicas, que están causadas por variaciones genéticas en uno o en un reducido número de genes6. Entre ellas, se han identificado los genes y las mutaciones causales de enfermedades como la hipercolesterolemia familiar7, el síndrome de QT largo, el síndrome de Brugada8 y algunas miocardiopatías9. El problema, desde el punto de vista de la salud pública, es que estas enfermedades suelen afectar a pocas personas, por lo que el impacto en la salud de la población suele ser modesto.
La identificación del componente genético de las enfermedades crónicas más frecuentes está siendo mucho más dificultosa ya que su progresión y manifestación clínica están relacionadas no sólo con factores genéticos, sino también con factores ambientales y su interacción. La cardiopatía isquémica (CI) es una de estas enfermedades que se caracteriza por diferentes formas de presentación clínica, una etiopatogenia compleja, su relación con factores ambientales (dieta, tabaco, actividad física)10, en la que, desde el punto de vista genético, intervienen muchos genes, cuyo efecto muchas veces puede estar modificado por factores ambientales. La CI continúa siendo la principal causa individual de muerte y las estimaciones indican que será la principal causa de muerte en el mundo en el año 202011. En España, la CI produjo el 9,98% de las muertes en 200612 y causa, además, una gran morbilidad13. Determinar sus bases genéticas podría tener un impacto relevante en la prevención y el tratamiento de esta enfermedad.
En esta revisión de carácter narrativo, tenemos como objetivo poner al día y sintetizar algunos de los resultados más relevantes obtenidos hasta la fecha sobre el estudio del componente genético de la CI. Para ello analizaremos las siguientes cuestiones: a) ¿hay un componente genético en la enfermedad? ¿podemos cuantificarlo?; b) ¿cuál es la arquitectura genética de la enfermedad?, es decir ¿qué genes y qué variantes genéticas influyen en su aparición?, y c) ¿cuál es la utilidad de esa información genética en la prevención, el diagnóstico y el tratamiento de la CI?
CUANTIFICACIÓN DEL COMPONENTE GENÉTICO DE LA CARDIOPATÍA ISQUÉMICADesde hace años existe el convencimiento de que los genes influían en el riesgo de presentar enfermedades del corazón14,15. Esta evidencia proviene de estudios epidemiológicos clásicos en los que se observaba que la aparición de diferentes formas clínicas de CI eran más frecuentes en individuos con antecedentes familiares de la enfermedad, lo que indicaba una agregación familiar14,16. Estudios más recientes indican que el antecedente de enfermedad isquémica en padres17 o hermanos18,19 es un factor de riesgo de aparición de CI independiente de los factores de riesgo clásicos. Esta agregación familiar apunta a un componente genético en la enfermedad, aunque también podría estar relacionada con factores ambientales y comportamientos que son comunes y también «se heredan» en el contexto familiar20.
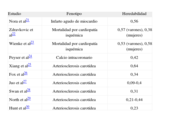
La heredabilidad se define como la proporción de la variabilidad fenotípica existente en una población que puede ser atribuible al componente genético. Se han publicado varios estudios que han determinado la heredabilidad de distintos fenotipos relacionados con la arteriosclerosis21–30, que indican, en su mayoría, que la heredabilidad es del 40-55%, lo cual apoya la influencia genética sustancial en estos fenotipos (tabla 1). De todos modos, hay que tener en cuenta que la interpretación del resultado de la heredabilidad se tiene que realizar con precaución31: los componentes genético y ambiental de la variabilidad de una característica poblacional no suelen ser independientes, y su efecto muchas veces depende de la interacción entre ellos. Esta interacción no suele ser tenida en cuenta cuando se calcula este estimador. La heredabilidad depende de la población en la que se estudia; por ejemplo, en una población con un estilo de vida muy similar el componente ambiental tendrá un impacto más pequeño en la variabilidad existente en la población que estará más relacionada con variabilidad genética y viceversa.
Estudios que han analizado la heredabilidad de distintos fenotipos relacionados con la arteriosclerosis
| Estudio | Fenotipo | Heredabilidad |
| Nora et al21 | Infarto agudo de miocardio | 0,56 |
| Zdravkovic et al22 | Mortalidad por cardiopatía isquémica | 0,57 (varones), 0,38 (mujeres) |
| Wienke et al23 | Mortalidad por cardiopatía isquémica | 0,53 (varones), 0,58 (mujeres) |
| Peyser et al24 | Calcio intracoronario | 0,42 |
| Xiang et al25 | Arteriosclerosis carotídea | 0,64 |
| Fox et al26 | Arteriosclerosis carotídea | 0,34 |
| Juo et al27 | Arteriosclerosis carotídea | 0,09-0,4 |
| Swan et al28 | Arteriosclerosis carotídea | 0,31 |
| North et al29 | Arteriosclerosis carotídea | 0,21-0,44 |
| Hunt et al30 | Arteriosclerosis carotídea | 0,23 |
Uno de los grandes retos de la investigación actual es determinar qué genes, y dentro de estos genes, qué variantes genéticas son las que causan las enfermedades complejas en general, y entre ellas, la CI en particular: en esto consiste la definición de la arquitectura genética. Para responder a este objetivo, principalmente, hay dos aproximaciones metodológicas: los estudios de ligamiento y los estudios de asociación. Dentro de estos últimos hay varios tipos de estudios que explicaremos a continuación.
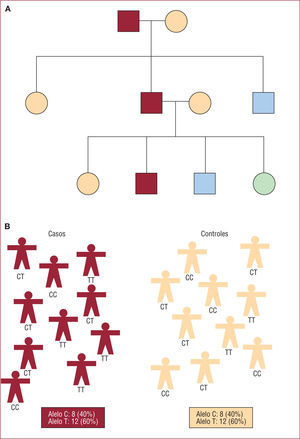
Estudios de ligamientoLos estudios de ligamiento permiten identificar zonas del genoma en las que se encuentra algún gen o genes que están asociados con la enfermedad en estudio. Este tipo de estudios requieren la participación de varias familias integradas por individuos afectos y no afectos de la enfermedad. Clásicamente se han utilizado unos 400 marcadores distribuidos de forma uniforme a lo largo del genoma32, y se ha analizado si alguno de estos marcadores se observaba más frecuentemente en los individuos afectos, es decir si su transmisión de generación en generación se relaciona con la aparición de la enfermedad (segregación). En la figura 1A se presenta un ejemplo en el que se observa que el alelo A1 de la variante A cosegrega con la enfermedad de forma autosómica dominante, es decir la presencia de un único alelo A1 ya implica la aparición de la enfermedad. Estos resultados no indican que el alelo A1 sea la variante genética que cause la enfermedad, sino que está asociada (en ligamiento) con la variante causal. El siguiente paso es analizar con mayor detalle la zona del genoma alrededor de ese alelo A1 con otra serie de marcadores genéticos para identificar el gen y la variante causal. Esta aproximación permitió identificar el gen causal de la enfermedad de Huntington en 198333 y ha sido muy útil para identificar los genes causales de muchas enfermedades monogénicas.
. B: representación del fundamento de los estudios de asociación en un estudio de caso-control que compara la frecuencia de alelos entre casos y controles.")
A: representación gráfica del fundamento de los estudios de ligamiento en la que se observa que la presencia del alelo A1 de la variante A cosegrega con la enfermedad de forma autosómica dominante (cuadrados representan sexo masculino; círculos representan sexo femenino; color marrón oscuro representa personas que presentan enfermedad). B: representación del fundamento de los estudios de asociación en un estudio de caso-control que compara la frecuencia de alelos entre casos y controles.
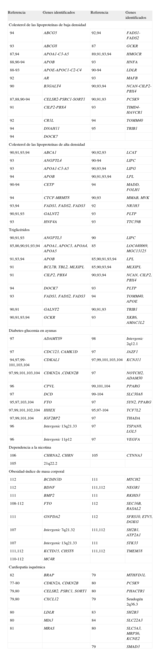
Sin embargo, la utilización de este tipo de estudios en enfermedades complejas como la CI no ha tenido tanto exito34. En la tabla 2 se presentan los resultados de diferentes estudios de ligamiento realizados en familias con CI35–42; se observa que el solapamiento o la concordancia de los locus identificados en los diferentes estudios han sido nulos. Esta falta de replicabilidad puede indicar una heterogeneidad genética en la enfermedad (muchos genes involucrados), aunque también puede estar relacionada con los diferentes fenotipos relacionados con la CI utilizados en los diferentes estudios. De los locus identificados en estos estudios, en únicamente dos se ha podido identificar el hipotético gen causal.
Resultados de los estudios de ligamiento realizados en familias con casos de cardiopatía isquémica (CI)
| Estudio | Población | Fenotipo | Locus identificado | Gen candidato en el locus |
| Pajukanta et al35 | Finlandia | 156 pacientes que tenían al menos 2 hermanos con CI (demostrada angiográficamente) prematura | Xq25 | AGTR2 |
| Francke et al36 | Isla Mauricio | 99 casos índice con CI antes de los 52 años y algún hermano con infarto de miocardio o diabetes | 16p13.3 | SOCS1, ACSM3 |
| Broeckel et al37 | Alemania | 513 casos índice con al menos 2 hermanos con CI antes de los 60 años | 14q32 | (?) |
| Wang et al38 | Estados Unidos (origen europeo) | 1 familia de 25 miembros con 13 afectos de CI | 15q26 | MEF2A |
| Wang et al39 | Estados Unidos (origen europeo) | 428 familias con al menos 2 hermanos con CI | 1p36 | GJA4 |
| Hauser et al40 | Estados Unidos (origen europeo) | 438 casos índice con al menos un hermano con CI | 3q13 | (?) |
| Helgadottir et al41 | Islandia | 296 familias con varios casos de infarto de miocardio | 13q12-13 | ALOX5AP |
| BHF Family42 | Gran Bretaña | 1.933 casos índice con al menos 2 hermanos con CI | 2q14.3-21.2 | IL1A, IL1B, PROC |
BHF: British Heart Foundation.
En el estudio de Wang et al39 se identificó, mediante el estudio de una familia, una mutación (deleción de 21 bases) en el gen MEF2A que segregaba de forma dominante con la enfermedad y los hallazgos se replicaban en una muestra de unos 200 casos y controles. Sin embargo, aunque hay un estudio en una serie española que ha replicado estos resultados43, la mayoría de los estudios no han confirmado esta asociación44,45 y el papel de este gen sigue siendo motivo de debate46.
Helgadottir et al41 utilizaron 1.068 marcadores genéticos, en 713 individuos de 296 familias y encontraron un locus que segregaba con infarto agudo de miocardio en el cromosoma 13, especialmente en mujeres41. Se analizó con mayor detalle ese locus mediante 120 marcadores en un grupo de unos 800 casos y 800 controles y se encontró, finalmente, un haplotipo en el gen ALOX5AP que conllevaba un exceso de riesgo de infarto de miocardio e ictus41. Estos resultados se han replicado en otras poblaciones y estudios47,48, pero no en todos49. Este gen codifica una proteína activadora de la lipooxigenasa 5, que interviene en la producción de leucotrienos, y que en modelos animales se ha relacionado con la arteriosclerosis50. Actualmente, se ha convertido en una nueva diana terapéutica en la vía inflamatoria de la arteriosclerosis51.
Estudios de asociaciónLos estudios de asociación permiten identificar variantes genéticas relacionadas con la enfermedad en estudio. Este tipo de estudios utilizan diseños clásicos en la investigación biomédica: caso-control o de cohorte, aunque también hay algún tipo de diseño específico para estudios genéticos que incluyen tríos definidos por un caso índice y sus padres52. Estos estudios ya se utilizaron en los años cincuenta para analizar la relación entre los grupos sanguíneos y el riesgo de úlcera péptica53. En los años noventa se hicieron muy populares y se ha publicado gran cantidad de estudios que analizan la relación entre variantes genéticas y distintos fenotipos. En la figura 1B se presenta el fundamento de estos estudios que se basa en determinar las diferencias en la frecuencia de genotipos o de alelos entre casos y controles. Recientemente se han publicado unas excelentes y extensas revisiones sobre cómo interpretar un estudio de asociación aplicado a la genética54–56.
Estudios clásicos de asociaciónEstos estudios se fundamentan en una hipótesis a priori y el conocimiento de la etiopatogenia de la enfermedad y seleccionan uno o varios genes candidatos y, dentro de este gen, una o varias características genéticas, y analizan su asociación con el fenotipo de interés. A pesar de la gran cantidad de estudios que han utilizado este abordaje para determinar la arquitectura genética de las enfermedades complejas, los resultados han sido decepcionantes. En una revisión publicada en 2002, que analizaba varias enfermedades complejas, se concluyó que había un reducido número de genes que hubieran demostrado tener una asociación sólida57. En el caso de la CI, en esta revisión57, se identificaron diferentes estudios que habían analizado la asociación entre marcadores genéticos de 35 genes y el riesgo de presentar CI. En estos estudios se había analizado 21 variantes genéticas estudiadas en tres o más ocasiones y ninguna de estas variantes se replicaba en más del 75% de los estudios57. La asociación entre la CI y el gen APOE es la que presenta resultados más consistentes58; se observa que, en comparación con los individuos con el genotipo E3/E3, los portadores del alelo E2 tienen un menor riesgo de CI, mientras que los portadores del alelo E4 tienen un exceso de riesgo [alelo ApoE2 = rs429358(T) + rs7412(T); alelo ApoE3 = rs429358(T) + rs7412(C); alelo ApoE4 = rs429358(C) + rs7412(C)].
Las razones del poco éxito que ha tenido este tipo de estudios son varias y se han discutido en varias revisiones57,59,60: a) la magnitud de la asociación entre los marcadores genéticos y las enfermedades complejas es pequeña (generalmente, odds ratio [OR] < 1,3); b) los estudios tienen poco poder estadístico debido al pequeño tamaño muestral; c) en los estudios con poder estadístico reducido las posibilidades de encontrar un resultado falso positivo aumentan; d) sesgo por estratificación de poblaciones, cuando se incluyen en un estudio casos y controles de distintas poblaciones en las que la prevalencia de la enfermedad y del alelo de riesgo en estudio son diferentes, se puede encontrar asociaciones espurias; e) normalmente la variante genética analizada no suele ser la causal, sino que suele estar en desequilibrio de ligamiento (asociada) con la variante genética causal, por este motivo la magnitud de la asociación de la variante analizada suele ser menor y, por lo tanto, también es menor el poder estadístico para detectarla como estadísticamente asociada; f) hay interacciones con otros marcadores genéticos y con el ambiente que modulan la magnitud de la asociación con el fenotipo de interés, y g) en muchas ocasiones la definición de la enfermedad es compleja y puede introducir heterogeneidad, por ejemplo y en el caso de la CI, la angina y el infarto agudo de miocardio son dos manifestaciones clínicas que pueden tener una arquitectura genética diferente.
Toda esta controversia sobre los estudios de asociación se ha traducido en la reciente publicación de unas recomendaciones sobre los aspectos a tener en cuenta cuando se realiza un estudio de asociación genética61 y cómo evaluar las pruebas que este tipo de estudios proporciona62, teniendo en cuenta el tamaño de muestra de los estudios, la replicación de los resultados en muestras independientes y los posibles sesgos.
Estudios de asociación con fenotipos extremosEstos estudios también se fundamentan en una hipótesis a priori y el conocimiento de la etiopatogenia de la enfermedad y generalmente se basan en la selección de un gen candidato y la secuenciación de este gen en personas que presentan características extremas de la distribución del fenotipo de interés. Por ejemplo, se podría seleccionar a individuos con concentraciones de colesterol > 280 y < 170mg/dl o presión arterial sistólica > 180 y < 110mmHg, o con infarto agudo de miocardio antes de los 45 años y sin infarto después de los 85 años. Teóricamente, estos individuos tan distintos presentarán características genéticas también diferentes. Se selecciona un gen candidato y se secuencia para identificar variantes genéticas en los individuos con el fenotipo de riesgo extremo y ausentes en los individuos con fenotipo de protección extrema. Si se identifican estas variantes, se pueden realizar estudios en modelos animales y celulares para demostrar su impacto funcional, y además se pueden realizar estudios en poblaciones para determinar la frecuencia de estas variantes y su asociación con el fenotipo en estudio.
Utilizando este tipo de aproximación, Cohen et al63 seleccionaron el gen PCSK9 y lo secuenciaron en 128 individuos con concentraciones muy bajas de lipoproteínas de baja densidad (LDL). Este gen codifica una proteasa que modula el número de receptores para la LDL hepáticos, un aumento de la actividad de esta proteasa reduce el número de receptores hepáticos y produce hipercolesterolemia en ratones. Se identificaron dos mutaciones (Y142X y rs28362286/C679X) que se asociaban con un importante descenso de los valores de LDL. Se han identificado más variantes genéticas en este gen, presentes entre el 0,2 y el 32% de la población que también se asocian con las concentraciones de LDL64. Posteriormente, el mismo grupo de investigadores demostró que estas variantes también se asociaban con un menor riesgo de acontecimientos coronarios65; estos resultados se han replicado en otros estudios66,67.
Nuestro grupo, mediante una aproximación similar, analizó la relación entre variantes genéticas en el gen KCNMB1 y el control de la presión arterial. Este gen codifica la subunidad β1 que regula la actividad de un canal de potasio que regula el tono vascular. Se secuenció este gen en 11 individuos con hipertensión severa y en 12 sujetos normotensos y se identificó una mutación (rs11739136/E65K) que aumentaba la funcionalidad del canal de potasio y conllevaba cifras más bajas de presión arterial diastólica68, esta variante genética también se asociaba con un menor riesgo de acontecimientos cardiovasculares69. Estos resultados se han replicado en otros estudios70,71 y también se ha observado que esta variante genética modula la respuesta a antagonistas del calcio72 y bloqueadores beta73.
Estudios de asociación integral del genoma (genome-wide association; GWA)Este tipo de estudios se propusieron a mediados de los años noventa74, no están basados en una hipótesis previa y se fundamentan en dos supuestos: a) hay un número de variantes genéticas que capturan gran parte de la variabilidad genética común conocida en el genoma, y b) las enfermedades raras o mendelianas están relacionadas con variantes genéticas poco frecuentes, mientras que las enfermedades crónicas están relacionadas con variantes genéticas frecuentes (hipótesis de enfermedad común en relación con variante genética común).
Durante la siguiente década se desarrollaron los instrumentos para poder realizar este tipo de estudios. Se constituyó un consorcio que fue recogiendo en una base de datos las variantes genéticas por cambio de secuencia del ADN (polimorfismos de base única o single nucleotide polimorphisms, SNP) identificadas (SNP Consortium)75, donde actualmente se han registrado más de diez millones de polimorfismos. Pero ¿es necesario genotipar los diez millones de polimorfismos para estudiar la genética de las enfermedades complejas? Para responder a esta pregunta el estudio HapMap ha genotipado más de tres millones de estos polimorfismos y ha determinado el patrón de desequilibrio de ligamiento existente en el genoma en distintas poblaciones4,5. Este estudio concluyó que al genotipar unos 500.000 polimorfismos se captura gran parte de la variabilidad existente en el genoma. En paralelo, diferentes compañías pusieron en marcha plataformas para el genotipado masivo y chips que permitían el genotipado de este número de variantes genéticas. En 2006 todo estaba listo para poner en marcha este tipo de estudios y responder a la pregunta de si variantes genéticas comunes explican una parte de la variabilidad de las enfermedades comunes76.
En los últimos 3 años se han publicado varios estudios de GWA que han analizado variantes genéticas asociadas con cardiopatía isquémica77–84 y sus factores de riesgo: variables lipídicas85–94, hipertensión95, diabetes o glucemia en ayunas94–104, dependencia a la nicotina105,106, obesidad o índice de masa corporal107–112. Los resultados más importantes de estos estudios se presentan en la tabla 3. Actualmente hay más estudios en marcha e iniciativas para realizar metaanálisis de los estudios previos que, sin duda, contribuirán a incrementar el conocimiento de las variantes genéticas relacionadas con estos fenotipos de interés. El National Human Genome Research Institute mantiene activa una página web donde van actualizando los resultados de este tipo de estudios (http://www.genome.gov/gwastudies/).
Genes asociados con cardiopatía isquémica y sus factores de riesgo identificados en estudios de asociación integral del genoma
| Referencia | Genes identificados | Referencia | Genes identificados |
| Colesterol de las lipoproteínas de baja densidad | |||
| 94 | ABCG5 | 92,94 | FADS1-FADS2 |
| 93 | ABCG8 | 87 | GCKR |
| 87,94 | APOA1-C3-A5 | 89,91,93,94 | HMGCR |
| 88,90-94 | APOB | 93 | HNFA |
| 88-93 | APOE-APOC1-C2-C4 | 90-94 | LDLR |
| 92 | AR | 93 | MAFB |
| 90 | B3GALT4 | 90,93,94 | NCAN-CILP2-PBX4 |
| 87,88,90-94 | CELSR2-PSRC1-SORT1 | 90,91,93 | PCSK9 |
| 91 | CILP2-PBX4 | 93 | TIMD4-HAVCR1 |
| 92 | CR1L | 94 | TOMM40 |
| 94 | DNAH11 | 95 | TRIB1 |
| 94 | DOCK7 | ||
| Colesterol de las lipoproteínas de alta densidad | |||
| 90,91,93,94 | ABCA1 | 90,92,93 | LCAT |
| 93 | ANGPTL4 | 90-94 | LIPC |
| 93 | APOA1-C3-A5 | 90,93,94 | LIPG |
| 94 | APOB | 90,91,93,94 | LPL |
| 90-94 | CETP | 94 | MADD, FOLH1 |
| 94 | CTCF-MRMT8 | 90,93 | MMAB, MVK |
| 93,94 | FADS1, FADS2, FADS3 | 92 | NR1H3 |
| 90,91,93 | GALNT2 | 93 | PLTP |
| 93 | HNF4A | 93 | TTC39B |
| Triglicéridos | |||
| 90,91,93 | ANGPTL3 | 90 | LIPC |
| 85,86,90,91,93,94 | APOA1, APOC3, APOA4, APOA5 | 85 | LOC440069, MGC13125 |
| 91,93,94 | APOB | 85,90,91,93,94 | LPL |
| 91 | BCL7B, TBL2, MLXIPL | 85,90,93,94 | MLXIPL |
| 91 | CILP2, PBX4 | 90,93,94 | NCAN, CILP2, PBX4 |
| 94 | DOCK7 | 93 | PLTP |
| 93 | FADS1, FADS2, FADS3 | 94 | TOMM40, APOE |
| 90,91 | GALNT2 | 90,91,93 | TRIB1 |
| 90,91,93,94 | GCKR | 93 | XKR6, AMAC1L2 |
| Diabetes-glucemia en ayunas | |||
| 97 | ADAMTS9 | 98 | Intergenic 2q12.1 |
| 97 | CDC123, CAMK1D | 97 | JAZF1 |
| 94,97,99-101,103,104 | CDKAL1 | 97,99,101,103,104 | KCNJ11 |
| 97,99,101,103,104 | CDKN2A ,CDKN2B | 97 | NOTCH2, ADAM30 |
| 96 | CPVL | 99,101,104 | PPARG |
| 97 | DCD | 99-104 | SLC30A8 |
| 95,97,103,104 | FTO | 97 | SYN2, PPARG |
| 97,99,101,102,104 | HHEX | 95,97-104 | TCF7L2 |
| 97,99,101,104 | IGF2BP2 | 97 | THADA |
| 96 | Intergenic 13q21.33 | 97 | TSPAN8, LGL5 |
| 96 | Intergenic 11p12 | 97 | VEGFA |
| Dependencia a la nicotina | |||
| 106 | CHRNA2, CHRN | 105 | CTNNA3 |
| 105 | 21q22.2 | ||
| Obesidad-índice de masa corporal | |||
| 112 | BCDIN3D | 111 | MTCH2 |
| 112 | BDNF | 111,112 | NEGR1 |
| 111 | BMP2 | 111 | RKHD3 |
| 108-112 | FTO | 112 | SEC16B, RASAL2 |
| 111 | GNPDA2 | 112 | SFRS10, ETV5, DGKG |
| 107 | Intergenic 7q21.32 | 111,112 | SH2B1, ATP2A1 |
| 107 | Intergenic 13q21.33 | 111 | STK33 |
| 111,112 | KCTD15, CHST8 | 111,112 | TMEM18 |
| 110-112 | MC4R | ||
| Cardiopatía isquémica | |||
| 82 | BRAP | 79 | MTHFD1L |
| 77-80 | CDKN2A, CDKN2B | 80 | PCSK9 |
| 79,80 | CELSR2, PSRC1, SORT1 | 80 | PHACTR1 |
| 79,80 | CXCL12 | 79 | Seudogén 2q36.3 |
| 80 | LDLR | 83 | SH2B3 |
| 80 | MIA3 | 84 | SLC22A3 |
| 81 | MRAS | 80 | SLC5A3, MRPS6, KCNE2 |
| 79 | SMAD3 | ||
En el caso de la CI, los resultados iniciales procedentes de 2 estudios publicados en junio de 2007, en el mismo número de la revista Science, sorprendieron al presentar el hallazgo común de una asociación de unos marcadores genéticos localizados en una zona intergénica del cromosoma 9 (próximos a los genes CDKN2A y CDKN2B) con el riesgo de CI77,78. Estos resultados fueron replicados a los pocos meses por el estudio del Wellcome Trust79 y, recientemente, en un estudio con participación española, esta zona también se observa como la más firmemente relacionada con el riesgo de infarto de miocardio80. El mecanismo funcional de esta asociación actualmente está siendo estudiado por varios grupos.
Los marcadores genéticos y genes identificados en relación con el riesgo de CI en los diferentes estudios de GWA pueden encontrarse en la tabla 3. La magnitud de la asociación entre las variantes genéticas identificadas y el riesgo de presentar CI es pequeña, con OR que oscilan entre 1,1 y 1,3. Otro de los resultados más sorprendentes de estos estudios es que la mayoría de los genes identificados no hubieran sido clasificados como genes candidatos a priori. Únicamente se habría incluido el LDLR y el PCSK9 en la lista de genes candidatos debido a su relevancia en el metabolismo lipídico. Los genes PSRC1, MIA3, SMAD3 y MRAS tienen como función regular el crecimiento celular79. El gen CXCL12 sintetiza unas citocinas que activan el proceso inflamatorio y se ha relacionado con angiogénesis y función endotelial113. El gen MTHFD1L codifica una sintetasa mitocondrial que regula la síntesis de la purina y la metionina y puede contribuir a las concentraciones de homocisteína plasmáticas. En conjunto, estos resultados apuntan a mecanismos conocidos (metabolismo lipídico, inflamación), pero también a nuevas vías para explorar relacionadas con la aparición y la progresión de la arteriosclerosis.
UTILIZACIÓN DE LA INFORMACIÓN GENÉTICA PARA PREVENCIÓN, DIAGNÓSTICO Y TRATAMIENTO DE LA CARDIOPATÍA ISQUÉMICAClásicamente, se ha afirmado que la identificación de marcadores genéticos asociados con la CI puede ser útil para mejorar su prevención, el tratamiento y el conocimiento sobre el mecanismo etiopatogénico de la arteriosclerosis.
Información genética y prevención de la cardiopatía isquémicaUna de las utilidades de los marcadores genéticos podría incluir la identificación de personas con un mayor riesgo de presentar CI y que serían susceptibles de estrategias de prevención más agresivas. La hipótesis es que la inclusión de variantes genéticas relacionadas con la CI en las funciones de riesgo mejoraría su validez, es decir, la capacidad de diferenciar a los individuos que presentarán la enfermedad de los que no (discriminación) y/o la reclasificación de los individuos en estratos de riesgo que incluya la información genética que se ajustaría más al riesgo observado.
La discriminación es la capacidad de un modelo predictivo de diferenciar a los individuos que sufren un episodio de enfermedad coronaria de los que no lo sufren. Generalmente, se cuantifica con el parámetro estadístico c, que es análogo al área bajo la curva ROC (receiver operative characteristics); este valor es una estimación de la probabilidad de que un modelo asigne un riesgo más alto a los individuos que presentan una enfermedad coronaria en un período de seguimiento de 5 años que a los que no la presentan114.
Hasta la actualidad se han publicado 3 estudios que han analizado si la incorporación de información genética en las funciones de riesgo clásicas mejora la capacidad predictiva y discriminatoria de estas funciones115–117. Morrison et al, con un seguimiento de más de 15.000 participantes del estudio ARIC, construyeron en esta cohorte una escala de riesgo genético que incluía 11 polimorfismos y que se basaba en el número de alelos de riesgo de cada individuo115. Aunque esta escala de riesgo genético se relacionaba de forma independiente con el riesgo de presentar CI en el seguimiento (en blancos el RR para presentar CI por cada incremento de 1 unidad de la puntuación genética fue 1,10, con un intervalo de confianza de 1,06-1,14), esta puntuación no mejoraba la capacidad de discriminación de los factores de riesgo clásicos (el área bajo la curva ROC pasaba de 0,764 a 0,766)115.
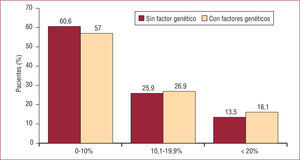
Kathiresan et al también utilizaron una puntuación de riesgo genético que incluía 9 polimorfismos relacionados con variables lipídicas, también basados en el número de alelos de riesgo de cada individuo116. Esta puntuación de riesgo genético se relacionaba de forma independiente con la aparición de acontecimientos cardiovasculares en la cohorte de Malmo. Aunque esta puntuación no mejoraba la capacidad de discriminación de los factores de riesgo clásicos (área bajo la curva ROC, 0,8 con y sin puntuación genética), sí que mejoraba la reclasificación de individuos de forma significativa. Al analizar a los participantes que presentaban acontecimientos cardiovasculares en el seguimiento, observaron que cuando se tenía en cuenta la información genética aumentaba el número de participantes clasificados de riesgo intermedio o alto respecto a las funciones de riesgo clásicas (fig. 2).
, e incorporando la información genética al cálculo del riesgo. Adaptado de Kathiresan et al116.")
Porcentaje de participantes que presentan un acontecimiento cardiovascular en el seguimiento, clasificados como de riesgo bajo, intermedio o alto según los criterios de la ATP-III (sin factores genéticos), e incorporando la información genética al cálculo del riesgo. Adaptado de Kathiresan et al116.
Recientemente, en el estudio de la cohorte de enfermeras americana, la incorporación de la variabilidad genética en el cromosoma 9p21.3 no mejoraba la capacidad de discriminación (área bajo la curva ROC de 0,807 a 0,809) ni la reclasificación de individuos respecto a los obtenidos con los factores de riesgo clásicos117.
Es necesario realizar nuevos estudios que incorporen todas las pruebas disponibles consolidadas hasta el momento, que provienen fundamentalmente de los estudios de GWA, para analizar si con este conjunto de información sobre las funciones de riesgo puede mejorar nuestra capacidad de identificar a los individuos con mayor riesgo.
Información genética y tratamiento de la cardiopatía isquémicaLos estudios genéticos pueden ayudar a identificar nuevas dianas terapéuticas al identificar nuevas vías relacionadas con la etiopatogenia de la CI.
Como ejemplo, tras la identificación de la asociación entre el gen ALOX5AP y la CI, se han desarrollado fármacos que inhiben la proteína que activa la lipooxigenasa 551, que han demostrado reducir el valor de algunos biomarcadores de mal pronóstico en pacientes con infarto agudo de miocardio portadores de variantes genéticas de riesgo en este gen.
Otro ejemplo claro es la identificación del gen PCSK9 y su relación no sólo con la hipercolesterolemia familiar, sino también con las concentraciones de LDL en la población general64 y con el riesgo de infarto agudo de miocardio65,66,80. Actualmente se están diseñando fármacos con esta diana terapéutica para el control de las concentraciones de colesterol y, quizá, la prevención de la CI118.
Otra área de investigación y desarrollo es la farmacogenómica, que tiene como objetivo identificar variantes genéticas que modulan la respuesta a fármacos. La Food and Drug Administration aprobó en 2007 una prueba genética para determinar la sensibilidad al tratamiento oral anticoagulante y ajustar la dosis inicial y de mantenimiento y la periodicidad de los controles analíticos119. En esta prueba se determinan variantes genéticas en dos genes: CYP2C9120,121 y VKORC1122,123, relacionados con la dosis de anticoagulante requerida y la aparición de complicaciones.
Recientemente, se han publicado 2 estudios en los que se analiza si las variantes genéticas en unos genes determinados modulan la eficacia del clopidogrel en pacientes con infarto agudo de miocardio124,125. La conclusión de estos estudios es que los portadores de determinadas variantes genéticas en el gen CYP2C19 tienen menores concentraciones del metabolito activo del clopidogrel, una menor inhibición de la actividad plaquetaria y una mayor tasa de acontecimientos cardiovasculares (muerte, reinfarto, ictus) al año de seguimiento, especialmente en el subgrupo de pacientes a los que se ha implantado un stent.
Información genética y etiopatogenia de la cardiopatía isquémicaComo hemos comentado anteriormente, los resultados disponibles sobre la arquitectura genética de la CI apuntan a mecanismos causales conocidos (metabolismo lipídico, inflamación), pero también a nuevas vías relacionadas especialmente con el control del crecimiento celular, que merecen ser exploradas.
El estudio de las asociaciones entre características genéticas y enfermedad puede ayudarnos a inferir las causas ambientales y genéticas de la enfermedad en estudio mediante lo que se ha llamado «aleatorización mendeliana»126. La aleatorización mendeliana se fundamenta en el hecho de que la información genética de un individuo se determina al azar en el momento de la formación de los gametos y la fecundación. El resultado es que la distribución poblacional de las variantes genéticas es independiente de factores ambientales y comportamientos que normalmente actúan como variables confusoras en los estudios epidemiológicos clásicos. El concepto de aleatorización mendeliana se puede utilizar para analizar si la asociación de algunos fenotipos intermedios, como el colesterol o la proteína C reactiva, con la CI son causales o no126. Como se representa en la figura 2, este análisis se fundamenta en determinar la relación entre los tres tipos de variables de interés: la variante genética, el fenotipo intermedio y la enfermedad. Si la variante genética se asocia con el fenotipo intermedio y éste, a su vez, se asocia con la enfermedad, si la asociación es causal, la variante genética se debe asociar con la enfermedad (fig. 3A). Si la variante genética no se asocia con la enfermedad, la asociación entre el fenotipo intermedio y la enfermedad puede deberse a que hay variables confusoras, causalidad reversa o atenuación de la magnitud de las asociaciones por errores en las medidas (fig. 3B).

Un ejemplo reciente lo tenemos con el análisis sobre la posible relación causal de la proteína C reactiva con la CI127. En este estudio se observó que determinadas variantes genéticas del gen de la proteína C reactiva se asociaban con las concentraciones plasmáticas de esta proteína, que a su vez se asociaban con la aparición de cardiopatía e ictus isquémico; sin embargo, no se observaba ninguna asociación entre las variantes genéticas analizadas y el riesgo de presentar un acontecimiento clínico isquémico, lo que cuestiona el papel causal de la proteína C reactiva en la CI127,128.
RETOS PARA EL FUTUROLas conclusiones más importantes que podemos extraer del conocimiento actual y el estado de la investigación genética son: que el esclarecimiento de la arquitectura genética de la CI no está siendo sencillo, que las variantes genéticas identificadas tienen un efecto modesto y que explican una parte pequeña de la variabilidad de los fenotipos analizados. Para continuar avanzando habrá que vencer varios retos que tendremos que afrontar de forma multidisciplinaria y a distintas escalas:
- –
Los estudios deberán tener un gran tamaño de muestra para tener suficiente poder estadístico para identificar asociaciones con un efecto pequeño (OR de 1,1 o cambios en la presión arterial o concentraciones de colesterol de 1mmHg o 1–1,5mg/dl, respectivamente). Para ello, será necesario seguir estableciendo consorcios y colaboraciones entre distintos grupos, como algunos que ya se han constituido a escala nacional e internacional. La creación y el mantenimiento de biobancos, como el Banco Nacional de ADN (www.bancoadn.org), que incluye un catálogo de diferentes enfermedades cardiovasculares, constituirán también elementos fundamentales de la estrategia.
- –
Los estudios realizados hasta ahora aún no han tenido en cuenta los factores ambientales y de estilos de vida y la interacción gen-ambiente en la modulación del riesgo: éste es un aspecto muy relevante129 que requiere una participación activa multidisciplinaria con médicos, epidemiólogos y, naturalmente, genetistas. Será necesario recoger información ambiental con cuestionarios validados para luego analizar este tipo de interacciones.
- –
Las variantes genéticas comunes identificadas explican una pequeña parte de la variabilidad de los fenotipos estudiados. La mayor parte de los estudios han analizado polimorfismos, variantes genéticas que implican un cambio de una base en la secuencia del ADN en un locus concreto, pero hay otro tipo de variantes poco estudiadas, como las variantes de número de copias (copy number variation), que pueden tener gran relevancia130. También el estudio de las modificaciones no estructurales del ADN, como puede ser la metilación de alguna de sus estructuras sin cambios en la secuencia, puede tener un impacto importante en la salud ya que puede regular la expresión de estos genes (epigenética).
- –
Desarrollar métodos para incorporar la información genética en la predicción del riesgo cardiovascular y validar su utilidad131,132.
- –
Si las variantes genéticas comunes no explican gran parte de la variabilidad de los fenotipos cardiovasculares de interés, no hay que descartar que haya variantes genéticas raras que influyan en estos fenotipos. Para identificar estas variantes se están poniendo en marcha estudios de secuenciación del genoma133.
- –
Toda esta información genética requerirá programas de formación y puesta al día para los profesionales sanitarios que tengan que usarla134.
- –
Por último, pero no menos importante, toda esta información se tiene que contextualizar en los aspectos ético, legal y social135.
Los autores declaran no tener conflicto de intereses.
Esta revisión ha sido financiada en parte por el Ministerio de Ciencia e Innovación, Instituto de Salud Carlos III (Red HERACLES RD06/0009).