

Nuestro objetivo es proporcionar al clínico los conocimientos básicos para la interpretación de datos referidos a enfermedades genéticas que le sirvan de ayuda a la hora de tomar decisiones clínicas. Se hace un símil del genoma humano con una enciclopedia, la «enciclopedia de la vida», analizando como se transmite la información genética, las causas, las consecuencias y las principales reglas de nomenclatura de las mutaciones. Se discute a continuación qué mutaciones y cómo pueden contribuir a la aparición de la enfermedad cardiovascular. Por último, se describe la contribución de la genética a la mejora del diagnóstico y el tratamiento de la hipercolesterolemia familiar (HF), una de las entidades en que el tratamiento hipolipemiante es eficaz y coste-efectivo. Se justifica el empleo de técnicas de detección genética a gran escala como son los biochips que analizan las principales mutaciones de los genes que originan el HF.
Palabras clave
El símil más utilizado para describir el genoma humano, cuya secuencia se publicó por primera vez en 20011,2, es el de una enciclopedia3. La “enciclopedia de la vida” consta de 44 volúmenes (dos copias de cada uno de los 22 cromosomas denominados autosomas) y dos volúmenes adicionales, los cromosomas sexuales (dos copias del X en las mujeres y XY en los varones) lo que hace un total de 46 tomos.
El alfabeto utilizado en el genoma humano tiene solamente 4 letras, o bases: adenina A, guanina G (purínicas), timina T y citosina C (primidínicas). El número total de letras del genoma humano (genoma diploide) es 3.000 millones y una parte de las letras se encuentran agrupadas en capítulos (los genes). Como si de un libro secreto se tratase, los capítulos no son uniformes. Las zonas que poseen «significado» (los exones, que suponen un 1-2% del total de la enciclopedia) están separadas por zonas que carecen de él (los intrones), que constituyen alrededor del 25% del total. Entre los capítulos hay grandes áreas (un 75% del total de la enciclopedia) cuyo significado se desconoce (zonas intergénicas); estas zonas son las más numerosas.
Las zonas de los capítulos de la enciclopedia que poseen «significado» (los exones) se traducen a otro lenguaje formado por veinte palabras diferentes (aminoácidos) que dan lugar a frases (las proteínas). Las palabras de este nuevo lenguaje son el resultado de traducir combinaciones de tres letras del lenguaje original (codones). Algunas combinaciones de tres letras codifican la misma palabra, por eso decimos que el código genético es degenerado. Existen señales mediante combinaciones de letras que indican dónde empiezan (ATG) las frases (codón de iniciación [start codon]) y tres combinaciones de letras (TGA, TAG y TAA) que marcan dónde terminan (codón de parada [stop codon]).
En la actualidad la enciclopedia no tiene un índice completo, es decir, no conocemos todos los genes, sino uno parcial. Algunos capítulos todavía no están localizados y, por lo tanto, carecen de un título. Como en una enciclopedia en la que algunos capítulos carecen de título, existen zonas que con gran probabilidad son genes pero cuyas identidad y función todavía son desconocidas. Nuestro genoma, según el Human Genome Project, podría tener unos 30.000 genes que podrían dar lugar a la síntesis de hasta 100.000 proteínas diferentes.
TRANSMISIÓN DE LA INFORMACIÓN GENÉTICACada capítulo de nuestra «enciclopedia» se copia y se transmite de célula a célula y de generación en generación. Durante la realización de las copias se pueden producir errores (mutaciones) en la secuencia del ADN, entre los que podemos encontrar pequeñas y grandes inserciones y deleciones. Estos errores se transmiten en las sucesivas reproducciones de la enciclopedia (transmisión a la descendencia)4. Aunque el 99,9% del genoma humano es idéntico en todos los individuos, hay cierta variabilidad como consecuencia de los procesos mencionados. Se produce aproximadamente un cambio de letra, lo que denominamos polimorfismo de un solo nucleótido (single nucleotide polymorphism [SNP]) cada 1.000 letras, pares de bases (pb). Por lo tanto, hay cerca de 3 millones de SNPs en todo el genoma que explicarían la mayor parte de la variabilidad interindividual.
Los SNP son, en gran medida, la causa de la variabilidad genética interindividual observada. Se pueden distinguir varios tipos de SNP: los rSNP (aleatorios [random]) son los que están situados en la zona silenciosa del genoma y representan el 90% del total; los gSNP (gen asociado), que podrían influir en el control de los genes, supondrían cerca de 1 millón, y los cSNP (de codón), que se encuentran en zonas codificantes y a menudo influyen en la función del gen. El SNP Consortium ha anotado 1,7 millones. Se ha podido relacionar algunos de estos cambios (se han anotado unos 80.000 en la base de datos de la Universidad de Cardiff: http://www.hgmd.cf.ac.uk) con alguna enfermedad, pero el efecto de la mayoría es totalmente desconocido e impredecible.
TRASCRIPCIÓN DE LA INFORMACIÓN GENÉTICAEl ácido desoxirribonucleico (ADN) genómico posee dos tipos de secuencias, las que llevan información para la síntesis de proteínas (genes) y las que no conducen a la síntesis de una molécula activa de ácido ribonucleico (ARN) o proteína (regiones intergénicas). Éstas podrían desempeñar funciones muy variadas: funciones estructurales en el cromosoma, empaquetamiento del ADN, de organización de la cromatina dentro del núcleo, o incluso de regulación de la expresión génica.
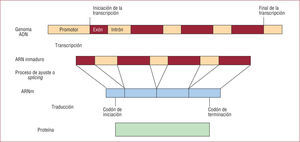
Por su parte, los genes dan lugar a los transcritos de ARN, un proceso en el que interviene la enzima ARN polimerasa. La molécula de ARN sintetizada puede tener sus propias funciones (ARN de transferencia [ARNt], ribosómicos [ARNr] y micro-ARN), pero en muchos casos es necesaria la traducción a proteína para que sea funcional. Como ya se ha comentado, la mayoría de los genes estructurales de los organismos eucariotas están organizados en regiones codificantes interrumpidas por secuencias no codificantes llamadas intrones (fig. 1).
, para producir una molécula de ARN mensajero (ARNm), que posteriormente se traducirá en una molecula de proteína en los ribosomas.")
En el proceso de transcripción, la ARN polimerasa II realiza una copia de ARN correspondiente al gen. Los intrones son eliminados del transcrito del ARN inmaduro durante un proceso denominado de ayuste (splicing), para producir una molécula de ARN mensajero (ARNm), que posteriormente se traducirá en una molecula de proteína en los ribosomas.
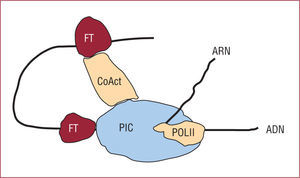
La transcripción es el proceso mediante el cual se sintetiza una molécula de ARN a partir de una secuencia complementaria de ADN. Los puntos correctos de inicio y final de la transcripción de un gen están identificados en el ADN molde por una secuencia promotora, situada corriente arriba del gen (extremo 5'), y una secuencia terminadora, situada corriente abajo del gen (extremo 3'). Las secuencias promotoras contienen regiones conservadas (denominadas cajas TATA, GC, CAAT, etc.) a las que se unen proteínas, denominadas factores de transcripción, que facilitan o impiden que la ARN polimerasa II inicie el proceso de transcripción (fig. 2). Además de estas secuencias promotoras, existen otras secuencias reguladoras situadas en la zona 5' del promotor, que permiten o facilitan la unión de moléculas que regulan la transcripción del gen. Algunos genes también poseen secuencias potenciadoras (enhancers) y/o atenuadoras, que pueden estar situadas tanto en la zona 5' como en la zona 3' del gen, e incluso en determinados casos en una región situada dentro del gen.
 —bien directamente, bien a través de coactivadores (CoAct)— estabilizan la unión del complejo de preiniciación (PIC), que incluye la ARN polimerasa II (POLII), al promotor del gen. La estabilización da lugar al comienzo de la síntesis del ARN.")
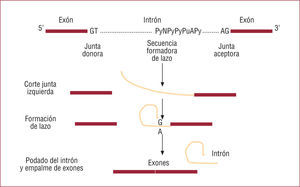
Una vez sintetizado el ARN, éste debe ser despojado de sus secuencias no codificantes (intrones) en el proceso de ayuste (splicing en la terminología inglesa) gracias a la existencia de secuencias conservadas5. Todo ello conduce a la formación del ARNm, que en los ribosomas es traducido a proteína (fig. 3).
 la secuencia donora está constituida por los nucleótidos GT y situada en la posición 5")
Proceso de ayuste: a) la secuencia donora está constituida por los nucleótidos GT y situada en la posición 5' del intrón; b) la secuencia aceptora, que está en el extremo 3' del intrón y está formada por el par AG, y c) la secuencia formadora de lazo, que está situada entre −18 y −40 pb del extremo 3' del intrón y tiene la secuencia PyNPyPyPuAPy (Py: base de pirimidina; N: cualquier base; Pu: base de purina). En el proceso de ayuste se forma un lazo entre el grupo fosfato de la posición 5' de la G (invariable de la junta izquierda del intrón) y el grupo OH de la posición 2' de la ribosa unida a la A (invariable de la secuencia formadora de lazo). Una vez que se ha formado el lazo, el paso siguiente es la eliminación del intrón y la unión de los extremos de los exones correspondientes para dar lugar al ARNm.
Las interacciones con agentes químicos (alteraciones químicas de la molécula de ADN como resultado de hidrolisis, oxidación o mutilación) o físicos (la luz ultravioleta es capaz de formar fotoproductos, como los dímeros de pirimidina entre bases de pirimidina adyacentes) pueden producir cambios en el ADN, pero la mayoría de ellos se producen de forma espontánea durante procesos celulares, tales como los que tienen lugar durante la replicación o la reparación del ADN6. En muchos casos estos cambios en el ADN crean desapareamientos de bases y dan lugar a mutaciones puntuales. Pero también pueden producirse cambios moleculares mayores, como deleciones e inserciones de diferente tamaño. Las deleciones o inserciones se producen cuando un fragmento de ADN se elimina o se inserta en un lugar específico del genoma.
Cuando una pequeña deleción o inserción tiene lugar en una región codificante, se obtiene como resultado una proteína mutada. En ocasiones puede mantenerse el marco de lectura de los codones (deleción o inserción en marco [inframe]), pero también puede ocurrir que dicho marco de lectura se vea modificado (deleción o inserción con cambio de marco [frameshift]) y se altere la secuencia proteínica corriente abajo de la mutación. Este tipo de mutaciones suele conducir a la aparición de un codón stop prematuro, por lo que se obtiene una proteína truncada.
Las mutaciones puntuales, según la zona del gen en que tienen lugar y dependiendo del tipo de mutación, pueden producir efectos muy variados en la funcionalidad de la proteína correspondiente. Las mutaciones que tienen lugar en las zonas promotoras, reguladoras o potenciadoras afectan a la expresión del gen si el nucleótido o los nucleótidos afectados son esenciales para la unión de un factor de transcripción. En cambio, las mutaciones situadas en la zona codificante de la proteína pueden: a) cambiar el marco de lectura, como hemos indicado, y dar como resultado una proteína con una secuencia diferente; b) modificar un solo aminoácido, en cuyo caso la proteína mutada es idéntica a la silvestre (wild type), con la excepción de que tiene un aminoácido cambiado (missense mutation); c) cambiar un codón que codifica un aminoácido por un codon stop (nonsense), en cuyo caso la proteína mutada será una proteína truncada, y d) cambiar un nucleótido que sea esencial para que el proceso de ayuste se efectúe de forma correcta, en cuyo caso se producirá un ARNm aberrante.
NOMENCLATURA DE LAS MUTACIONES: REGLAS BÁSICASHay cierta confusión en la terminología. El término mutación se utiliza en ocasiones para indicar un cambio y en otras para indicar un cambio que causa cierta enfermedad. De forma similar, el término polimorfismo se utiliza tanto para describir un cambio que no causa enfermedad como para señalar un cambio que se encuentra con una frecuencia ≥ 1% de la población. Para evitar esa confusión, es preferible no utilizar los términos mutación y polimorfismo (incluso SNP) y emplear términos tales como variaciones de secuencia, alteración o variante alélica7,8.
Según el tipo de nucleótido sustituido, las mutaciones puntuales se denominan: a) transición, que consiste en la sustitución de una pirimidina por otra o de una purina por otra, de tal forma que un par G-C es reemplazado por un par A-T o viceversa, y b) transversión, cuando una purina es reemplazada por una pirimidina o viceversa, en cuyo caso un par A-T es reemplazado por un par T-A o C-G.
Para nombrar las mutaciones se recomienda utilizar una serie de reglas elaboradas por el Human Genome Organization (HUGO) Gene Nomenclature Committee (HGNC), que se detallan en su página web (www.hgvs.org/mutnomen/recs) y resumimos a continuación:
- –
La regla más importante es que las variantes deben ser descritas en el nivel más básico posible, es decir a nivel de ADN. La descripción siempre debe referirse a una secuencia de referencia, ya sea la secuencia genómica o la codificante de ADN.
- –
Para genes/proteínas, sólo se utilizarán los símbolos recomendados por el HGNC (disponibles en: www.genenames.org/guidelines.html). Cuando se hace referencia a nivel de ARN o proteína en un determinado texto, la primera vez que se menciona debe utilizarse el siguiente formato: en primer lugar ADN y entre paréntesis proteína o ARN. Ejemplo: c.48G>C (p.Trp16Cys).
- –
Cuando se describen varios cambios, es conveniente proporcionar una tabla con la lista de los hallazgos, utilizando columnas separadas para ADN, ARN y proteínas, e indicar si los cambios fueron deducidos de forma experimental o teórica.
- –
Para hacer referencia a un nucleótido, deberá precederle la letra g en minúscula si se trata del ADN genómico, la c minúscula si se refiere a un ADN codificante (ADN-c), la m minúscula si se refiere a un ADN mitocondrial, la r minúscula si se refiere a una secuencia de ARN ribosomal y la p minúscula si se refiere a una proteína. Para evitar posibles confusiones entre el número del nucleótido y la g, la c, etc., se intercalará un punto. Por ejemplo g.576, significa que se trata del nucleótido que está situado en la posición 576 del ADN genómico.
En la numeración de nucleótidos de la secuencia de ADN de referencia, la A del codón de iniciación (ATG) se considera el nucleótido +1. No hay ningún nucleótido que se enumere con el cero. Los nucleótidos que se encuentran en posición corriente arriba del +1 deben ir precedidos por el signo menos; por ejemplo, –1, –2, –3, etc. Los nucleótidos situados corriente abajo del +1 no van precedidos de ningún signo.
La secuencia de referencia que se utilice para el ADN debe ser preferentemente la descrita en RefSeq database, que se encuentra en: www.ncbi. nlm.nih.gov/RefSeq/
En las secuencias de referencia genómicas: a) la numeración de nucleótidos es puramente arbitraria y empieza en el 1 del nucleótido de la base de datos de la secuencia de referencia. No se utilizan los signos + y –. Cuando no se conoce la secuencia genómica completa, se debe utilizar una secuencia de ADN codificante de referencia.
Para la descripción de cambios específicos:
- –
El símbolo > indica una sustitución a nivel de ADN; por ejemplo, c.75A>T.
- –
El símbolo _ indica un cambio que afecta a varios residuos, que separa el primero y el último de los residuos afectados; por ejemplo, c.77_79delACT.
- –
Con del se indica una deleción; por ejemplo, c.77delA.
- –
Con dup se indica una duplicación; por ejemplo, c.dupA.
- –
Con ins se indica una inserción; por ejemplo, c.75_76insG.
Los cambios de nucleótido deben empezar por el número del nucleótido seguido del cambio que tiene lugar en esa posición; por ejemplo, 576G>C significa que el nucleótido de la posición 576 que en la secuencia de referencia es una Gha sido reemplazado por una C.
Al describir dos variantes de secuencia en un mismo individuo:
- –
Dos cambios en la secuencia en diferentes alelos se representan entre corchetes, separados por el signo +; por ejemplo, c.[76A>C]+[87delG].
- –
Dos variantes de secuencia en el mismo alelo se ponen entre corchetes separados por punto y coma; por ejemplo, c.[76A>C; 83G>C].
- –
Dos variantes de secuencia en alelos desconocidos se ponen entre corchetes separadas por (+); por ejemplo, c.[76A>C(+)83G>C].
Para designar la localización de una variación en un intrón, se puede utilizar la numeración de los nucleótidos correspondientes al ADN-c. Por ejemplo, c.1232+1G>C significa la sustitución de G por C del primer nucleótido del intrón que sigue a la posición 1232 del ADN-c. Cuando no se dispone de la secuencia completa del ADN genómico para designar las mutaciones que tienen lugar en los intrones, se antepone IVS seguido del número del intrón y un número positivo, que corresponde a la posición del nucleótido a partir de la G de la junta donora (GT), o un número negativo, que corresponde a la posición del nucleótido a partir de la G de la junta aceptora (AG). Así, por ejemplo, IVS4+1G>C significa la sustitución de G por C del nucleótido +1 del intrón 4.
Descripciones a nivel de proteínaLas descripciones de variantes a nivel de proteína deben reflejar los cambios observados a nivel de proteína y no tratar de incorporar ningún conocimiento relativo a cambios a nivel de ADN.
El código de tres letras debe ser el preferido para describir los aminoácidos (tabla 1).
Códigos aminoácidos de tres y una letras
| Alanina | Ala | A |
| Arginina | Arg | R |
| Asparagina | Asn | N |
| Aspartato | Asp | D |
| Cisteina | Cys | C |
| Fenilalanina | Phe | F |
| Glicina | Gly | G |
| Glutamato | Glu | E |
| Glutamina | Gln | Q |
| Histidina | His | H |
| Isoleucina | Ile | I |
| Leucina | Leu | L |
| Lisina | Lys | K |
| Metionina | Met | M |
| Prolina | Pro | P |
| Serina | Ser | S |
| Tirosina | Tyr | Y |
| Treonina | THr | T |
| Triptófano | Trp | W |
| Valina | Val | V |
| Codón de parada | Ter | X |
La descripción cambio de marco de lectura (frameshift) no debe incluir la deleción del sitio donde está localizado dicho cambio del extremo C-terminal natural (codon stop) de la proteína; es decir, p.Arg97ProfsX23 y no p.97_Pro109delfsX23. La misma recomendación para las inserciones en marco, es decir, no describir los aminoácidos insertados, sólo la longitud total del nuevo fragmento; por ejemplo, p.Glu5ValfsX5 y no p. Glu5Valins2fsX3.
Para la nomenclatura de mutaciones basada en aminoácidos, el codón de iniciación metionina es el codón +1.
Las secuencias de referencia de la proteína deben ser representativas del producto de traducción primario, no de una proteína procesada, y por lo tanto han de incluir toda la secuencia del péptido señal.
Los aminoácidos originados por cambios introducidos corriente arriba de la iniciación de la traducción son numerados como los nucleótidos; por ejemplo, Ser-3, Thr-2.
Los aminoácidos originados por cambios resultantes en la traducción de secuencias intrónicas deben ser numerados como los nucleótidos; por ejemplo, Phe5-3, Gln5-2, Cys4+1.
Los aminoácidos originados por la desaparición de un codon stop que producen que la traducción se prolongue se nombran Arg*1, Ser*2, etc.
La X se utiliza para describir un codón de finalización de la traducción.
Para describir una sustitución, el aminoácido silvestre (wild type, el de mayor frecuencia en la población) se nombra antes y el mutado después; por ejemplo, Arg370Ser.
LA ENFERMEDAD CARDIOVASCULAR COMO UNA ENFERMEDAD GENÉTICA COMPLEJA. IDENTIFICACIÓN DE VARIANTES GÉNICAS QUE LA ORIGINANEn las enfermedades monogénicas casi todos los poseedores de un alelo padecen la enfermedad (se habla entonces de penetrancia elevada), lo que facilita la identificación de la variante génica que las causa. Ese no es el caso de la enfermedad cardiovascular (EC). Es cierto que existen tipos monogénicos, como la hipercolesterolemia familiar (HF) (véase más adelante), pero en general la enfermedad no sigue un patrón mendeliano típico. Algunos autores señalan que en el proceso podrían participar al menos un centenar de genes9, cuyo efecto dependería, además de determinadas condiciones ambientales, de un patrón característico de las enfermedades comunes.
La hipótesis denominada «enfermedades comunes, variantes comunes» propone que las variantes que causan las enfermedades comunes tienen elevada frecuencia en la población general10. Aunque se trata de la hipótesis con más apoyo, actualmente un número importante de científicos respaldan una alternativa en la que se incluirían también las combinaciones de variantes raras11. En ambas, sin embargo, se propone que el efecto aislado de cada una de las variantes debe de ser muy pequeño. Es la combinación de muchas variantes patogénicas lo que incrementaría la probabilidad de padecer la enfermedad. Así, en la población habría un continuo en uno de cuyos extremos estarían los poseedores de las variantes más patogénicas, que padecerán la enfermedad incluso cuando su estilo de vida sea saludable. En el otro extremo estarían los poseedores de las variantes saludables que pueden eludir la enfermedad incluso si su estilo de vida se aleja del aconsejado. Por lo tanto, la futura identificación de las variantes que causan la enfermedad podría tener un importante valor predictivo.
Como se ha comentado, existen millones de variantes en el genoma, de las cuales sólo unos pocos cientos estarían implicadas en el desarrollo de la EC. Los grandes estudios del genoma completo permiten el análisis simultáneo de más de medio millón de marcadores genéticos distribuidos por todo el genoma en grupos de individuos que presentan la enfermedad y su comparación con grupos de control. Aunque estos estudios integrales han revelado asociaciones importantes12, el análisis de tantas variantes supone una gran limitación estadística, de manera que sólo las asociaciones muy fuertes (con valores de p ≈ 10-8) se aceptan como estadísticamente significativos en el análisis13.
Una estrategia complementaria es la de los genes candidatos, en la que se buscan variantes asociadas a determinados genes. Si, por ejemplo, se sabe que el riesgo cardiovascular aumenta en presencia de concentraciones elevadas de colesterol o presiones sanguíneas altas, los genes que codifican proteínas que tengan un papel reconocido en estos procesos constituirían un objeto de estudio obvio. Como es lógico, la selección de los genes a estudiar exige un conocimiento previo de su papel y/o de la etiología de la enfermedad.
LAS VARIANTES EN LAS REGIONES REGULADORAS COMO CANDIDATAS IDEALESLas variantes situadas en las regiones codificantes normalmente producen efectos muy graves y no encajan en el modelo actual de enfermedades comunes. De ahí que la atención de los investigadores haya derivado hacia las variantes en las regiones reguladoras. Hay dos razones por las que este tipo de variantes resultan idóneas para explicar las enfermedades comunes. La regulación de la expresión génica se lleva a cabo sobre todo en la iniciación de la transcripción. Aquí tiene lugar la unión de un complejo multiproteínico (complejo de preiniciación [PIC]) en el promotor del gen para así comenzar la síntesis de ARNm. Tanto el acceso de las proteínas al ADN como la estabilidad del complejo formado son los principales factores que determinan la cantidad de ARNm sintetizada. En ambos procesos los factores de transcripción desempeñan un papel muy relevante. Llevan a cabo su función uniéndose a sus secuencias de reconocimiento en la región reguladora. Estas secuencias son específicas para cada uno de los factores y los cambios de secuencia pueden dar lugar a cambios en la afinidad del factor o incluso producir la aparición de un nuevo elemento regulador. En las secuencias de reconocimiento hay un elevado grado de flexibilidad y muy raramente la secuencia pierde totalmente su capacidad de unirse al factor como consecuencia del cambio de un nucleótido. Esta flexibilidad es la que puede servir para explicar la baja penetrancia de las mutaciones y lo que las hace ideales para explicar la base genética de las enfermedades comunes.
En general, este tipo de enfermedades no se hace evidente hasta que concurren ciertos factores ambientales. En casos como éste, es mucho más sencillo explicar la genética de la enfermedad si suponemos un modelo basado en la existencia de una variante en un elemento regulador. Hay que tener en cuenta que no todos los factores de transcripción están activados en la célula en todo momento. De hecho, la síntesis y/o la activación de los factores de transcripción son los principales sistemas por los que la célula responde a las señales del ambiente. Por otra parte, algunos factores de transcripción se expresan de forma exclusiva en algunos periodos del desarrollo y otros solamente se expresan o son activos ante determinado estímulo, sea éste químico u hormonal, algo también característico de enfermedades como la EC.
EJEMPLO DE LA HIPERCOLESTEROLEMIA FAMILIARLa HF es una enfermedad autosómica codominante caracterizada por concentraciones muy elevadas de colesterol de las lipoproteínas de baja densidad (cLDL) en sangre, aumento del riesgo de enfermedad coronaria prematura y depósitos extravasculares de lípidos como son los xantomas tendinosos y el arco corneal en las primeras décadas de la vida14. La HF se produce por mutaciones en el gen que codifica el receptor celular de las LDL (LDLR). Su ligando natural es la apoliproteína B (apoB), que es la proteína mayoritaria de las LDL. La HF es una enfermedad frecuente, ya que en la mayoría de los países la prevalencia de HF se sitúa en 1/500 sujetos en su forma heterocigota, por lo que se calcula que hay cerca de 100.000 sujetos afectos en España15.
La HF es la causa más frecuente (70%) y mejor conocida de las hipercolesterolemias autosómico-dominantes (HAD) caracterizadas por la presentación familiar de hipercolesterolemia con un claro patrón bimodal en las concentraciones de cLDL entre sus miembros16. Otras causas menos frecuentes (5%) de HAD son: a) las mutaciones en el gen de apoB, y entonces la HAD se denomina apoB-100 defectuosa familiar; b) mutaciones con ganancia de función en PCSK9 (< 1%), proteasa que está involucrada en la degradación del LDLR y se denomina FH3, y c) la hiperlipoproteinemia(a), que es la causa de HAD en aproximadamente un 2-3% de los casos17. Aproximadamente, en un 30% de las HAD se desconoce el defecto genético que las origina, aunque en algunas de ellas un aumento en la absorción intestinal de esteroles pudiera desempeñar un papel patogénico importante18. Los fenotipos lipídico y cardiovascular de las HAD dependientes del LDLR y apoB son indistinguibles entre sí y más graves que para el resto de las HAD19, por lo que su manejo clínico suele abordarse de forma conjunta y bajo la denominación HF20.
La HF es un ejemplo paradigmático de aterosclerosis precoz in vivo dependiente del cLDL y una de las entidades en las que el tratamiento hipolipemiante es más eficaz y coste-efectivo. La prevalencia de enfermedad coronaria en los sujetos heterocigotos HF antes de la era de las estatinas se situaba en torno al 50% de los varones y el 20% de las mujeres antes de los 50 años. En la actualidad todas las series muestran un descenso importante en la morbilidad y mortalidad cardiovascular debido al uso mucho más extensivo en estos pacientes de estatinas en monoterapia o en combinación con otros fármacos hipolipemiantes como ezetimiba21.
Diagnóstico clínico de la HFDado que disponemos de un tratamiento muy eficaz, el diagnóstico precoz de la HF es una prioridad sociosanitaria, como ha señalado la OMS22. El diagnóstico de la HF se ha venido realizando tradicionalmente según criterios clínicos basados en la presencia familiar y personal de enfermedad coronaria prematura, concentraciones de cLDL en sangre y la presencia de depósitos superficies de colesterol en el arco corneal y los xantomas tendinosos23–25. Sin embargo, el diagnóstico clínico presenta problemas: las concentraciones de colesterol total y cLDL no discriminan con suficiente sensibilidad a los sujetos afectos en la población general e incluso en el seno de una familia con el diagnóstico de HF. El solapamiento entre afectos y no afectos es especialmente importante en personas jóvenes, entre quienes aproximadamente el 20% tienen cifras de cLDL que pueden dar un diagnóstico equivocado26. Asimismo, el arco corneal y los xantomas son muy raros en los heterocigotos en las primeras décadas de la vida, por lo que ayudan poco en el diagnóstico, especialmente en jóvenes. Por estos motivos, los criterios clínicos empleados en el momento actual presentan una escasa correlación con el diagnóstico genético27. Nosotros hemos evaluado recientemente el valor de los tres criterios más frecuentemente utilizados en el diagnóstico clínico de la FH, y su sensibilidad y especificidad se encuentran en torno al 70%19, por lo que recomendamos realizar el diagnóstico genético en los casos descritos en la tabla 2.
Indicaciones para el diagnóstico genético con la sospecha de HF*
|
cLDL: colesterol de las lipoproteínas de baja densidad; HF: hipercolesterolemia familiar.
Se conocen más de mil mutaciones diferentes en LDLR y al menos tres en APOB causantes de HF, aunque la mutación CGG/CAG en el codón 3500 que sustituye glutamina por arginina (R3500Q) es la más frecuente causa de apoB defectuosa familiar. Esta gran heterogeneidad molecular es constante en la mayor parte de las poblaciones, incluida España, donde se conocen más de 250 mutaciones diferentes15. Los defectos son variados: desde mutaciones puntuales o pequeñas deleciones o inserciones (< 20 pb) a grandes reordenamientos que afectan a una gran parte del LDLR. Solamente en determinadas poblaciones con elevado aislamiento genético existen mutaciones con efecto fundador que hacen que muy pocas mutaciones originen HF y que la frecuencia de la enfermedad sea más alta de lo encontrado en otras poblaciones. Esto ocurre con la población franco-canadiense, los libaneses cristianos, los finlandeses, los judíos asquenazíes o población blanca de Sudáfrica14,16.
La confirmación de una mutación funcional en los genes del LDLR o apoB es de elección en la HF porque proporciona un diagnóstico de certeza. La utilidad del diagnóstico genético ha sido confirmada en muchos estudios, ya que identifica precoz e inequívocamente a sujetos con riesgo cardiovascular elevado, facilita el consejo genético, estratifica mejor el pronóstico de acuerdo con el tipo de mutación28, ayuda a la búsqueda de familiares afectos, facilita al médico la indicación de tratamiento y a los pacientes, su cumplimiento29, y en nuestro país, en muchos casos, permite a los pacientes acceder a una aportación reducida en la compra de las estatinas.
El análisis genético no puede aplicarse todavía a grandes grupos de población debido su coste, su disponibilidad y su complejidad, por lo que actualmente la principal recomendación de las guías internacionales para el manejo clínico de la HF es el diagnóstico genético en las siguientes situaciones20:
- 1.
Poblaciones en que unas pocas mutaciones causan la mayor parte de los casos de HF.
- 2.
Poblaciones en que la mayor parte de las mutaciones son conocidas y existen herramientas de diagnóstico genético rápido.
- 3.
Sujetos con fenotipos lipídicos equívocos en el seno de familias con mutación causante de la HF conocida.
El hecho de que al menos tres genes y no menos de varios centenares de mutaciones puedan ser la causa de una HF en la mayor parte de los sujetos obliga a emplear técnicas de detección genética a gran escala. Básicamente existen dos procedimientos: a) la secuenciación completa del LDLR, y b) el uso de microarrays o biochips de ADN. El del LDLR es un gen grande, por lo que su secuenciación y su análisis son costosos y laboriosos. Los microarrays consisten en un soporte donde se encuentran depositadas sondas de ADN que incluyen las mutaciones causales de la HF en una determinada población. La hibridación del ADN a estudiar permite la identificación de las mutaciones de forma rápida y con sensibilidad y especificidad > 99%. Su principal inconveniente es que requiere el conocimiento previo de las mutaciones de la HF en la población a la que se va a aplicar el test. Afortunadamente, en España se ha realizado un laborioso análisis de LDLR y APOB en la HF15, por lo que se ha optimizado un microarray que comprende 234 mutaciones en LDLR, las mutaciones de APOB y las principales mutaciones causales de hipercolesterolemia en PCSK930. Este microarray, denominado Lipochip, es pionero en el mundo en el diagnóstico de una enfermedad genética, su uso se ha generalizado en España y se está desarrollando en otros países con base genética conocida de la HF como Países Bajos, Italia y Estados Unidos.
CONCLUSIONESPara entender la estructura y cómo funciona el genoma humano, se puede comparar con una enciclopedia. Para nombrar las mutaciones genéticas se debe seguir unas reglas de nomenclatura aceptadas por la comunidad científica. Se puede considerar que la enfermedad cardiovascular es una enfermedad genética compleja, consecuencia de la interacción entre factores tanto genéticos como ambientales. La hipercolesterolemia familiar es una enfermedad autosómica codominante, ejemplo paradigmático de aterosclerosis precoz cuyo diagnóstico constituye una prioridad sociosanitaria, ya que se puede disminuir el riesgo cardiovascular de los pacientes mediante tratamientos que son eficaces.
Declaración de conflicto de interesesLos autores declaran no tener conflicto de intereses.
Este trabajo ha sido realizado en parte gracias a las ayudas de los proyectos: FIS PI06/0365, PI06/1068, RTIC C06/01 (RECAVA), CIBERER y SAF2005-07042.