La relación fundamental entre los trastornos sanguíneos y el sistema cardiovascular tiene su origen en múltiples puntos de contacto, que van del corazón y sus componentes estructurales, como las cámaras cardiacas, las válvulas, las arterias coronarias y las venas coronarias, a los vasos sanguíneos cerebrovasculares y periféricos. Mientras que los componentes celulares de la sangre circulante proceden inicialmente de células progenitoras pluripotenciales, los componentes plasmáticos, que incluyen las proteínas de la coagulación, se originan principalmente en la síntesis hepática y las células endoteliales. Presentamos aquí una revisión centrada en los trastornos hematológicos no oncológicos y sus posibles repercusiones en el sistema circulatorio arterial en forma de fenotipos frecuentes, como el infarto de miocardio, el ictus isquémico y los episodios de oclusión arterial periférica. En nuestro análisis utilizamos la tromboembolia venosa como patrón de referencia clínico. Presentamos también unos pasos prácticos y una guía para las pruebas diagnósticas y el manejo en la práctica clínica habitual.
Palabras clave
La sangre es el medio a través del cual se produce el intercambio de oxígeno, nutrientes y productos de desecho en todo el organismo y está formada por plasma, células hemáticas (hematíes, leucocitos) y plaquetas. Las plaquetas desempeñan un papel importante en la coagulación, los leucocitos originan la inflamación y los hematíes se encargan del transporte de oxígeno y nutrientes a todos los tejidos del organismo y retiran de los órganos los productos de desecho. Cualquier anomalía de estos componentes puede conducir a un trastorno hematológico. Mientras que los trastornos de las plaquetas y la coagulación que pueden conducir a la trombosis o la hemorragia constituyen uno de los principales campos de interés de la mayoría de cardiólogos, los trastornos de los hematíes y las plaquetas pueden afectar también a la mecánica del flujo sanguíneo y a la viscosidad de la sangre. Nuestro conocimiento de los trastornos hematológicos ha avanzado de manera constante en los últimos años con el desarrollo de la genética y las técnicas de biología molecular. Presentamos aquí una revisión centrada en los trastornos hematológicos no oncológicos y sus posibles repercusiones en el sistema circulatorio arterial en forma de fenotipos frecuentes, como el infarto de miocardio, el ictus isquémico y los episodios de oclusión arterial periférica. La tromboembolia venosa (TEV) se utiliza como patrón de referencia clínico para resaltar la apreciación de un problema frecuente al que se enfrentan todos los clínicos y diferenciar los trastornos sanguíneos que son específicos de los sistemas circulatorios venoso y arterial. Se presentan también pasos prácticos y una guía general para las pruebas diagnósticas y el manejo en la práctica clínica habitual.
Trastornos sanguíneos de la coagulaciónLa TEV es un problema de salud pública importante y creciente. Se estima que cada año en Estados Unidos 900.000 pacientes presentan una TEV clínicamente evidente y que ello da lugar a 300.000 muertes anuales por embolia pulmonar1. Los cardiólogos clínicos tienen la urgente tarea de conocer la carga que supone esta enfermedad y las posibles causas de la TEV, dado su enorme potencial de prevención y reducción de la morbimortalidad por esta causa.
Trombofilias hereditariasEn 1995, la Organización Mundial de la Salud y la International Society of Thrombosis and Hemostasis definieron la trombofilia como una tendencia anormal a la trombosis, que se caracteriza por manifestaciones como la edad de inicio temprana, episodios recurrentes, antecedentes familiares notables, localizaciones poco habituales, migratorias o generalizadas y una gravedad desproporcionada respecto a todo estímulo identificado. También hace referencia a los estados de hipercoagulabilidad que son el resultado final de enfermedades, trastornos o alteraciones que potencian la propensión del individuo a que se le formen coágulos de sangre en los sistemas venoso, arterial o microcirculatorio. Las características principales de las trombofilias hereditarias frecuentes se resumen en la Tabla 1.
Tabla 1. Resumen de las principales características de las trombofilias hereditarias
| Enfermedades | Mutaciones | Prevalencia | Manifestación | ||
| TEV | TEVR | TA | |||
| DAT | Gen SERPINC1 | 1/500 ∼ 1/5.000 | + | + | − |
| DPC | Gen de la proteína C | 1/200 ∼ 1/500 | + | + | +/− |
| DPS | PROS1 | 0,03-2% | + | + | +/− |
| Resistencia a APC | FV Leiden (A506G); FVR2 (H1299R); FV Cambridge (R306T) y FV Hong Kong (R306G) | 4-6% | + | + | − |
| Protrombina G20210A | Protrombina G20210A | 2-4% | + | +/− | − |
−: ausencia de evidencia; +: el riesgo aumenta; APC: proteína C activada; DAT: déficit de antitrombina; DPC: déficit de proteína C; DPS: déficit de proteína S; FV: factor V; TA: trombosis arterial; TEV: tromboembolia venosa; TEVR: tromboembolia venosa recurrente.
La antitrombina es una glucoproteína de cadena única que forma parte de la superfamilia del inhibidor de serina proteasa (serpina) y desempeña un papel anticoagulante clave mediante la prevención de la coagulación inadecuada, excesiva o en una localización errónea, que puede dar lugar a trastornos trombóticos. Hasta la fecha, se han descrito hasta 228 mutaciones diferentes en el gen SERPINC1 que se asocian a un déficit de antitrombina2, con una prevalencia descrita del déficit de antitrombina de entre 1/500 y 1/5.000 personas de la población general3. El déficit de antitrombina se asocia a un aumento del riesgo de embolia pulmonar y de TEV profunda de las extremidades superiores o inferiores, pero la TEV puede producirse también en localizaciones poco habituales, como las venas o los senos cerebrales o las venas mesentéricas, porta, hepáticas, renales y retinianas. Un metaanálisis de estudios observacionales ha descrito que el déficit de antitrombina aumenta significativamente el riesgo de una primera TEV (odds ratio [OR]=8,73; intervalo de confianza [IC] del 95%, 3,12-24,42) y de TEV recurrente (OR=3,37; IC del 95%, 1,57-7,2)4. A pesar de la clara relación con la TEV, que se inicia ya en la primera década de la vida, no hay ninguna evidencia clara de que el déficit de antitrombina aumente el riesgo de trombosis arterial5, 6.
Déficit de proteína CLa proteína C es una glucoproteína dependiente de la vitamina K sintetizada por el hígado. La trombina que forma complejos con la trombomodulina de las células endoteliales rompe un enlace de arginina-leucina dentro de la proteína C, lo que causa su activación. A su vez, la proteína C activada inactiva los factores Va y XIIIa; estos dos factores afectan a la vía de la coagulación activada por contacto y del factor tisular. La proteína C activada neutraliza también el inhibidor del activador del plasminógeno I, con lo que se potencia la actividad fibrinolítica. Se han descrito más de 160 mutaciones del gen de la proteína C7, y el déficit de proteína C congénito se transmite mediante un patrón autosómico dominante. La prevalencia descrita del déficit de proteína C asintomático es de entre 1/200 y 1/500 individuos sanos, mientras que la prevalencia del déficit de proteína C clínicamente significativo se ha estimado en 1/20.000 pacientes8.
El fenotipo clínico del déficit de proteína C heterocigoto simple puede ir del estado asintomático a la TEV grave. Se estima que un 50% de los portadores sufren una trombosis venosa antes de los 45 años de edad9. Un metaanálisis ha indicado que el déficit de proteína C causó un aumento significativo del riesgo de una primera TEV (OR=7,75; IC del 95%, 4,48-13,38) y el de TEV recurrente (OR=2,53; IC del 95%, 1,3-4,92)4. Además de la TEV, los pacientes heterocigotos para el déficit de proteína C pueden tener también riesgo de sufrir ictus isquémicos arteriales o trombosis de arterias mesentéricas. Otra revisión sistemática ha descrito una OR combinada de 6,49 (2,96-14,27) para el ictus isquémico arterial en los pacientes con déficit de proteína C5. Sin embargo, continúa sin haber evidencia alguna de una asociación entre los signos tempranos de alteraciones ateroscleróticas (grosor íntima-media, índice de presión tobillo/brazo) y el déficit de proteína C6. Teniendo en cuenta el conjunto de datos existentes, el déficit de proteína C puede ser un factor de riesgo de ictus isquémico arterial, especialmente en los pacientes de menos de 55 años de edad.
Déficit de proteína SLa proteína S es una glucoproteína dependiente de la vitamina K que se encuentra en un 40% en forma no ligada activa en la circulación y que actúa como principal cofactor de la proteína C activada, al aumentar la afinidad de la proteína por los fosfolípidos con carga eléctrica negativa. El complejo resultante de proteína C-proteína S activado, ligado a la membrana, produce un notable aumento de la inactivación del factor Va y el factor VIIIa. El déficit de proteína S hereditario es un trastorno autosómico dominante con casi 200 mutaciones diferentes del PROS1 identificadas que causan una pérdida de la función10, 11.
Aunque el déficit de proteína S es infrecuente en la población general, se encuentra en aproximadamente un 2% de los pacientes no seleccionados y en un 1-13% de los pacientes con trombofilia que sufren una TEV respectivamente12. Los individuos con déficit de proteína C presentan mayor riesgo de TEV y de TEV recurrente. Un metaanálisis ha puesto de manifiesto que el déficit de proteína S aumentó significativamente el riesgo de una primera TEV (OR=5,77; IC del 95%, 3,07-10,85) y de TEV recurrente (OR=3,76; IC del 95%, 1,76-8,04)4. En una amplia cohorte de familias con déficit de proteína S hereditario, la incidencia anual de TEV recurrente fue del 8,4% (IC del 95%, 5,8%-11,7%)13. Los resultados obtenidos en un amplio estudio de cohorte de familias indicaron también que los individuos con déficit de proteína S tienen un riesgo superior (hazard ratio [HR]=4,6; IC del 95%, 1,1-18,3) de trombosis arterial antes de los 55 años de edad14. Sin embargo, también hay estudios que no muestran ninguna evidencia convincente de que haya relación entre el déficit de proteína S y la trombosis arterial5, 6.
Resistencia a la proteína C activadaLa proteína C activada ejerce efectos anticoagulantes que van más allá de la inactivación del factor Va, mediante una rotura en R306 y R506, con lo que se genera factor V inactivo (FVai). Además, la proteína C activada genera una molécula anticoagulante (FVac) al romper el factor V (FV) de longitud completa en R506; esta FVac actúa como cofactor con la proteína C activada para degradar el factor VIIIa. La mutación más frecuente en el gen FV es una mutación puntual que causa una sustitución de Arg506 por Gln en uno de los lugares de rotura de la proteína C activada. Esta mutación, el FV Leiden, explica un 90-95% de todas las causas de resistencia a la proteína C activada15 y constituye la trombofilia hereditaria de mayor prevalencia, al afectar a un 4-6% de la población general16. Es muy poco frecuente o inexistente en poblaciones del Extremo Oriente, negros africanos o poblaciones autóctonas de América y Australia17. Las mutaciones menos frecuentes del FV afectan también a la resistencia a la proteína C activada. De ellas, la FVR2 (H1299R) reduce la actividad del cofactor de proteína C activada y da lugar a un aumento del riesgo trombótico cuando está presente en individuos heterocigotos compuestos con FV Leiden18. El FV Cambridge (R306T) y el FV Hong Kong (R306G) son mutaciones muy poco frecuentes que sólo muestran una resistencia leve a la proteína C activada y que no se han asociado a un aumento del riesgo de trombosis19.
La heterocigosis para FV Leiden produce un estado de hipercoagulabilidad de por vida, con un aumento de aproximadamente 3-7 veces del riesgo de trombosis venosa, y predice también la TEV recurrente4, 16. Además de la embolia pulmonar y la TEV profunda, el FV Leiden aumenta significativamente el riesgo de una primera trombosis venosa cerebral (OR=3,38; IC del 95%, 2,27-5,05)20. Se considera que el FV Leiden es un factor de riesgo moderado de ictus isquémico e infarto de miocardio antes de los 55 años, en especial en mujeres fumadoras. No se considera que sea un factor de riesgo de episodios de oclusión arterial periférica, excepto en algunos pacientes con una enfermedad nativa avanzada o una enfermedad de un injerto quirúrgico5, 6, 16.
Mutación génica G20210A del factor II (protrombina)El G20210A de la protrombina es una mutación de ganancia de función situada en la región 3’ no traducida del gen de la protrombina (nucleótido 20210 G a A), que da lugar a un aumento de las concentraciones plasmáticas de protrombina y a un estado de hipercoagulabilidad. El G20210A de protrombina es en frecuencia el segundo factor de riesgo hereditario de TEV. Su prevalencia depende de la ubicación geográfica y los antecedentes étnicos. Se da en un 2-4% de los individuos sanos del sur de Europa, lo cual es el doble de su frecuencia en los países del norte de Europa. Como el FV Leiden, es muy poco frecuente en las personas del Extremo Oriente, África y población autóctona de Australia y América. En la literatura, la presencia de la mutación se describe en un 6-8% de los pacientes con TEV21.
La mutación G20210A de la protrombina se asocia a un aumento de 2 a 3 veces en el riesgo de TEV4 y se asocia también a la TEV recurrente4, 22. Tal como ocurre con la mutación del factor V Leiden, la homocigosis se asocia al máximo riesgo general23. No se ha observado una asociación uniforme del G20210A de la protrombina con la trombosis arterial; sin embargo, es probable que haya un moderado aumento del riesgo en los individuos afectados de edad inferior a 55 años5.
Trombofilias adquiridasLos más frecuentes factores adquiridos que predisponen a la trombosis son la presencia de anticuerpos antifosfolipídicos y la enfermedad maligna. Se revisará aquí el síndrome antifosfolipídico. Otras causas menos frecuentes son los trastornos mieloproliferativos, el síndrome nefrótico, la hemoglobinuria paroxística nocturna y las etiologías iatrogénicas, como la quimioterapia contra el cáncer, la terapia hormonal para la infertilidad y las intervenciones quirúrgicas.
Síndrome antifosfolipídicoEl síndrome antifosfolipídico es un trastorno autoinmunitario caracterizado por la asociación clínica de anticuerpos antifosfolipídicos con un estado de hipercoagulabilidad y comporta un riesgo elevado tanto de TEV como de trombosis arterial24. El síndrome de anticuerpos antifosfolipídicos se asocia a infarto de miocardio, trombosis intracardiaca e hipertensión pulmonar que causa una predisposición a la trombosis y, con menor frecuencia, valvulopatías cardiacas y aterosclerosis de arterias periféricas y coronarias. Esto último podría explicarse por los efectos proinflamatorios y procoagulantes mediados por anticuerpos que se ejercen directamente en las células endoteliales25. Los pacientes con síndrome antifosfolipídico presentan una evolución cardiovascular significativamente peor26.
Estrategias diagnósticas para la trombosis venosa y arterialCuando se produce una trombosis venosa o arterial y se plantea la sospecha de una trombofilia adquirida, suelen realizarse pruebas de detección de anticoagulante lúpico. Los criterios diagnósticos actuales del síndrome antifosfolipídico exigen una prueba positiva en el panel de anticuerpos antifosfolipídicos (anticoagulante lúpico, título moderado o alto de anticuerpos anticardiolipina o anticuerpos anti-b2GP1) en dos ocasiones distintas con al menos 12 semanas de separación, en el contexto de una trombosis o complicaciones del embarazo27. Generalmente está indicado un estudio diagnóstico para detectar la trombofilia hereditaria tan sólo en los pacientes con antecedentes de múltiples episodios tromboembólicos, tromboembolia a una edad temprana, antecedentes familiares de tromboembolia, trombosis en una localización poco habitual o TEV sin ningún factor de riesgo evidente. El diagnóstico de las trombofilias hereditarias puede abordarse utilizando los siguientes pasos: a) deben realizarse ensayos amidolíticos funcionales en cada una de las trombofilias hereditarias antes citadas, excepto la mutación génica G20210A de la protrombina, que puede evaluarse directamente mediante un análisis genético; b) se llevan a cabo pruebas funcionales para descartar un déficit de antitrombina, déficit de proteína C o déficit de proteína S, y c) son necesarias pruebas genéticas para la detección de mutaciones génicas específicas como el FV Leiden. Es preferible evitar los análisis funcionales en el contexto de la trombosis aguda, y es mejor llevarlos a cabo antes o varios días después de suspender la administración de heparina o anticoagulación oral, puesto que la trombosis aguda y el tratamiento de anticoagulación pueden afectar a los resultados. Sin embargo, pueden realizarse análisis genéticos para detectar la mutación G20210A o el FV Leiden en cualquier momento. Es preciso descartar todas las causas de trombofilias adquiridas antes de clasificar a un paciente con un resultado anormal de las pruebas como afectado por una trombofilia hereditaria. Las pruebas a realizar en los pacientes con trombosis arterial, incluidos los fenotipos comunes de infarto de miocardio, ictus isquémico o episodios de oclusión arterial periférica, deben individualizarse prestando atención principalmente a los factores de riesgo tradicionales, las enfermedades sistémicas asociadas a la enfermedad vascular aterosclerótica y el síndrome antifosfolipídico con o sin un anticoagulante lúpico circulante, antes de iniciar pruebas para la identificación de trombofilias hereditarias en pacientes de menos de 55 años.
Estrategias de prevención y tratamiento de la trombosis venosa y arterialIndividuos asintomáticos: no se recomienda una profilaxis antitrombótica anticoagulante a largo plazo en los pacientes asintomáticos con trombofilias hereditarias, dado el aumento del riesgo de hemorragia26.
Profilaxis en contextos de alto riesgo: debe considerarse claramente la conveniencia de utilizar heparina o heparina de bajo peso molecular para la profilaxis antitrombótica cuando los pacientes con trombofilias hereditarias y trombosis arterial previa se encuentran en contextos de alto riesgo, como cirugía mayor, traumatismos o manejo del embarazo y el parto28.
Tratamiento de la trombosis venosa y arterial: el manejo inicial de la trombosis arterial coronaria en pacientes con una trombofilia hereditaria debe aplicarse siguiendo la asistencia estándar, con anticoagulantes y terapia dirigida a las plaquetas según esté indicado. El empleo de un tratamiento anticoagulante a largo plazo debe individualizarse, dada la ausencia de estudios aleatorizados. El manejo de la TEV no suele ser diferente del de este trastorno en otros pacientes sin trombofilias hereditarias28.
La piedra angular del manejo de las trombofilias adquiridas es el tratamiento de la enfermedad subyacente. Los pacientes con una positividad persistente de anticuerpos antifosfolipídicos y con antecedentes documentados de TEV o de tromboembolia arterial presentan un aumento del riesgo de recurrencia. El tratamiento anticoagulante a largo plazo es la base del tratamiento, con un objetivo de razón normalizada internacional de 2-3. No hay consenso general sobre el tratamiento profiláctico de los portadores de anticuerpos antifosfolipídicos que no han sufrido manifestaciones vasculares/trombóticas u obstétricas29. La profilaxis con ácido acetilsalicílico puede ser suficiente en los contextos de riesgo bajo.
Trastornos de las plaquetasTrombocitopenia inducida por heparinaLa trombocitopenia inducida por heparina (TIH), una reacción inmunitaria en respuesta a los complejos de factor plaquetario 4-heparina, pueden producirse en un 0,1-5% de los pacientes tratados con heparina, en función de la población de pacientes, el origen y la formulación de la heparina, la dosis y la duración del tratamiento. La TIH se produce con más frecuencia en los pacientes quirúrgicos que en los médicos y es más frecuente con la heparina no fraccionada que con la heparina de bajo peso molecular30. La TIH se caracteriza por trombocitopenia y una fuerte propensión a la trombosis paradójica, que se manifiestan en forma de trombosis arterial, microvascular o TEV, que es la más frecuente31, 32.
La TIH se diagnostica con el empleo de una combinación de criterios clínicos y de laboratorio. Hay dos criterios principales que son esenciales para establecer un diagnóstico clínico: aparición de trombocitopenia y/o trombosis clínica en relación temporal con el tratamiento de heparina (es característico que se produzca en un plazo de 5-14 días tras la exposición a heparina)33 y haber descartado otras causas de trombocitopenia. La detección de anticuerpos de TIH es necesaria para el diagnóstico, pero no suficiente, ya que solamente un subgrupo de los individuos que desarrollan anticuerpos para la heparina presenta realmente una TIH. Disponemos de varios análisis para detectar los anticuerpos de la TIH, como el de liberación de serotonina, el inmunoanálisis de absorción ligado a enzimas o el inmunoanálisis enzimático.
La suspensión de la heparina y el inicio de un anticoagulante alternativo deben ser medidas inmediatas en cuanto se plantea la sospecha clínica e incluso antes de que se disponga del resultado de ninguna de las pruebas serológicas34. Los anticoagulantes alternativos que pueden utilizarse son los inhibidores directos de trombina lepirudina, argatrobán y bivalirudina y el agente anti-Xa fondaparinux34. El empleo de anticoagulación con warfarina se ha asociado a la aparición de gangrena venosa de extremidades y debe evitarse hasta que haya mejorado el recuento de plaquetas. Además, debe iniciarse sin anticoagulación no heparínica concurrente y sin administración en bolo34. En los pacientes con TIH que no han sufrido un episodio tromboembólico, la anticoagulación terapéutica debe continuarse hasta que el recuento de plaquetas se haya estabilizado de nuevo34. Los pacientes con TIH que presentan una complicación tromboembólica deben recibir una tanda estándar de anticoagulación terapéutica para el episodio clínico específico34. La declaración de consenso del American College of Chest Physicians recomienda un seguimiento de los recuentos de plaquetas con determinaciones cada 2 o 3 días, empezando al cuarto día tras el inicio del tratamiento, hasta que este se interrumpa o hasta el día 14 de exposición a heparina34.
Púrpura trombocitopénica trombóticaLa púrpura trombocitopénica trombótica (PTT) es un trastorno multisistémico con peligro para la vida, causado por el depósito de plaquetas y de factor von Willebrand en las arteriolas y capilares que, a su vez, origina una isquemia generalizada de órganos, que afecta sobre todo al cerebro, el corazón y los riñones. Se produce entonces una anemia hemolítica microangiopática cuando los hematíes pasan por los vasos afectados y se rompen, lo que da lugar a unos fragmentos denominados esquistocitos35. La PTT, un trastorno muy poco frecuente debido al déficit de ADAMTS13, afecta a aproximadamente 1.000 personas al año en Estados Unidos, tiene una prevalencia mucho más alta en las mujeres, y su incidencia en afroamericanos es 9 veces superior a la existente en personas de otras etnias36. El conjunto de las cinco manifestaciones clásicas de trombocitopenia, anemia hemolítica, fiebre, disfunción renal y síntomas neurológicos, se da tan sólo en una minoría de los pacientes con PTT37. En un estudio de un solo centro se observó que el 21% de los pacientes con PTT presentaron un infarto de miocardio incidente38. En los pacientes cardiovasculares, el uso de antiagregantes plaquetarios como clopidogrel y ticlopidina se ha asociado excepcionalmente a una PTT inducida por la medicación. La PTT asociada a clopidogrel se produce a menudo en un plazo de 2 semanas tras el inicio de la administración. La tasa de supervivencia en los pacientes con PTT asociada a clopidogrel es de aproximadamente el 71,2%39.
El cuadro analítico es muy similar al de la coagulación intravascular diseminada, aunque el tiempo de protrombina y el tiempo de tromboplastina parcial activado rara vez son anormales40. Los análisis para determinar la actividad de ADAMTS13 han supuesto un avance en el diagnóstico de la PTT, con sensibilidades del 89-100% y una especificidad superior al 91%41. La plasmaféresis o la infusión de plasma son la base del tratamiento de la PTT. Se han utilizado glucocorticoides, ciclosporina, vincristina, esplenectomía y, más recientemente, rituximab (anticuerpo monoclonal para el CD20) en combinación con plasmaféresis, para tratar la PTT, aunque al respecto no hay datos de ensayos clínicos aleatorizados42. Por otra parte, algunos autores han señalado la utilidad del uso terapéutico de antiagregantes plaquetarios conjuntamente con la plasmaféresis, a pesar de la trombocitopenia significativa42. De forma análoga, una norma de tratamiento estándar anterior establecía que las transfusiones de plaquetas estaban contraindicadas en la PTT, pero una revisión publicada recientemente ha llegado a la conclusión de que continúa sin estar claro si esta medida es nociva o no43.
Trombocitopenia inmunitariaLa trombocitopenia inmunitaria (TPI) es un trastorno adquirido, de mecanismo inmunitario, que se caracteriza por una trombocitopenia aislada (recuento de plaquetas 150×109/l), sin que haya otras causas de trombocitopenia. Aunque la aparición de autoanticuerpos contra las glucoproteínas plaquetarias continúa teniendo un papel central en la fisiopatología de la TPI, se han identificado diversas anomalías que afectan a los mecanismos celulares de la modulación inmunitaria44. Parece que tanto la supervivencia como la producción de las plaquetas están alteradas en la TPI.
Un análisis de la base de datos General Practice Research Database del Reino Unido45 indicó que la estimación actual de la incidencia de la TPI es de 3,3/105 individuos adultos/año. Feudjo-Tepie et al46 han descrito prevalencias de TPI en adultos y población general de 23,6/100.000 y 20,3/100.000 en Estados Unidos en los años 2002-2006. Los signos y síntomas son muy diversos y van del paciente completamente asintomático a la hemorragia manifiesta en cualquier localización, de las que la más grave es la intracraneal. Aunque el recuento de plaquetas sea bajo, en algunos pacientes puede producirse un infarto de miocardio o un ictus isquémico47.
El diagnóstico de TPI se hace descartando otras causas de trombocitopenia. El enfoque diagnóstico básico para la TPI incluye la historia clínica, exploración física, hemograma completo, detección de anticuerpos antiplaquetarios y examen de un frotis de sangre periférica. En los pacientes de más de 60 años, es apropiada una punción de médula ósea para descartar la leucemia, las enfermedades infiltrativas y las anemias aplásicas.
El tratamiento de los pacientes con TPI debe tener en cuenta la edad del paciente, la gravedad de la enfermedad y la evolución natural prevista. Los pacientes adultos y en especial los de edad superior a 60 años tienen una incidencia de hemorragias mayores o mortales superior a la de los niños. Sin embargo, puede no ser necesario un tratamiento específico a menos que el recuento de plaquetas sea < 20×109/l o haya una hemorragia extensa. De hecho, el tratamiento actual de la TPI se considera apropiado para los pacientes sintomáticos y los que presentan riesgo de hemorragia48. Siempre que la situación del paciente no comporte un riesgo para la vida, los corticoides constituyen el tratamiento inicial estándar de la TPI. Generalmente se recomienda el empleo de inmunoglobulina intravenosa para los pacientes que no responden a los corticoides. El recuento de plaquetas se puede reforzar con el empleo de inmunoglobulina anti-D, activa solamente en pacientes con Rh positivo y en el contexto previo a la esplenectomía48. La esplenectomía se ha considerado tradicionalmente el tratamiento de segunda línea en los adultos con TPI en los que no se ha conseguido alcanzar un valor seguro del recuento de plaquetas con la terapia inicial de prednisona y es eficaz en la mayoría de los casos. En los últimos años se han producido avances en el tratamiento de la TPI crónica. Dichos avances incluyen la incorporación del tratamiento inmunomodulador (rituximab, anticuerpo monoclonal anti-CD20) y el desarrollo de agentes estimuladores de la trombopoyesis (romiplostim, eltrombopag), que se han utilizado en ensayos clínicos y han mostrado un efecto beneficioso importante49, aunque se desconocen sus posibles repercusiones negativas a largo plazo.
Tras las intervenciones coronarias percutáneas, los pacientes con TPI presentan riesgo de hemorragia o complicaciones trombóticas cuando se les administra o se deja de usar, respectivamente, el tratamiento antiagregante plaquetario. Dada la escasez de datos sobre la TPI en relación con la implantación de stents, no pueden hacerse recomendaciones estrictas y el tratamiento deberá individualizarse para reducir al mínimo el riesgo tanto de hemorragia como de trombosis. De todos modos, varios casos indican la factibilidad de la revascularización percutánea en pacientes seleccionados con enfermedad coronaria de múltiples vasos y TPI47.
Anomalías de los hematíesAnemiaLa Organización Mundial de la Salud define la anemia como una concentración de hemoglobina < 13g/dl en los varones y < 12g/dl en las mujeres posmenopáusicas. La anemia es frecuente en los pacientes con enfermedad cardiovascular. La prevalencia de anemia en los pacientes con insuficiencia cardiaca congestiva oscila entre el 4 y el 61% (mediana, 18%) según cuál sea la población estudiada, y la mayoría de los estudios indican una prevalencia > 20%50. La anemia parece tener mayor prevalencia en los pacientes de edad avanzada y en los que tienen limitaciones graves de la capacidad funcional y una mayor gravedad de la comorbilidad de enfermedad renal50. En los pacientes con infarto de miocardio, se observó anemia en situación basal en el 10,5% de los casos51 y la incidencia de anemia adquirida en el hospital fue elevada (un 33-69% según el centro)52.
Las etiologías de la anemia son multifactoriales e incluyen la pérdida hemática en el contexto de uso de tratamientos antiagregantes plaquetarios o anticoagulantes, las ferropenias, la hemodilución, la activación de la cascada inflamatoria, las pérdidas urinarias de eritropoyetina y la insuficiencia renal asociada52, 53. La anemia es un potente predictor independiente del aumento de mortalidad, hospitalización y riesgo de hemorragia en los pacientes con enfermedad cardiovascular51, 52, 53.
El tratamiento y el manejo de la anemia se han centrado en el empleo de agentes estimuladores de la eritropoyetina y suplementos de hierro por vía parenteral. Múltiples estudios indican un efecto positivo de la eritropoyetina o sus derivados cuando se administran en combinación con hierro oral o intravenoso, con mejoras de la función sistólica y diastólica ventricular izquierda y derecha, la dilatación y la hipertrofia, así como de la función renal54. Es necesario un ensayo aleatorizado de fase III con la potencia adecuada en este campo. El estudio STAMINA-HeFT, un ensayo multicéntrico, a doble ciego, aleatorizado y controlado con placebo, mostró una tendencia no significativa a un menor riesgo de mortalidad por cualquier causa o de una primera hospitalización por insuficiencia cardiaca en los pacientes tratados con darbepoetina alfa, en comparación con placebo (HR=0,68; IC del 95%, 0,43-1,08; p=0,1)55. Sin embargo, el ensayo Trial to Reduce Cardiovascular Events with Aranesp Therapy (TREAT) indicó que el uso de darbepoetina alfa en pacientes con diabetes mellitus, enfermedad renal crónica y anemia moderada no dializados no redujo el riesgo de la variable de valoración combinada primaria (es decir, muerte o evento cardiovascular o muerte o evento renal) y se asoció a un aumento del riesgo de ictus56. En consecuencia, no se ha establecido el umbral óptimo para iniciar el tratamiento ni el grado de corrección que se considera seguro y deseable para un paciente con insuficiencia cardiaca concreto. Actualmente se está llevando a cabo el segundo ensayo de mortalidad y morbilidad, el Reduction of Events with Darbepoetin alfa in Heart Failure (RED-HF), que es probable que aporte más respuestas57. Más recientemente, el estudio Reduction of Infarct Expansion and Ventricular Remodeling With Erythropoietin After Large Myocardial Infarction (REVEAL), un ensayo multicéntrico, aleatorizado, a doble ciego y controlado con placebo llevado a cabo por Rao et al, está evaluando los efectos de la epoetina α en el tamaño del infarto y el remodelado ventricular izquierdo en pacientes con infartos de miocardio grandes. Sus resultados, presentados en las Sesiones Científicas de 2010 de la American Heart Association celebradas en Chicago, indican que una inyección intravenosa de eritropoyetina tras una intervención coronaria percutánea primaria o de rescate practicada con éxito no redujo el tamaño del infarto en pacientes con infarto agudo de miocardio con elevación del segmento ST58. La evidencia existente indica que el uso sistemático de transfusiones o de agentes estimuladores de la médula ósea no aporta un efecto beneficioso y, de hecho, puede ser nocivo en los pacientes con infarto de miocardio o insuficiencia cardiaca, incluso en presencia de una insuficiencia renal concomitante59.
Enfermedad de células falciformesLa enfermedad de células falciformes (ECF) es un trastorno genético hereditario que se caracteriza por una anomalía de la hemoglobina denominada hemoglobina S (HbS). Incluye un grupo de anemias hemolíticas en las que la HbS está presente en forma homocigótica (HbSS) o heterocigótica compuesta, como cuando se combina con hemoglobina C (HbSC) o talasemia β (HbS-talasemia β). La ECF se produce por la homocigosis para una mutación de un único nucleótido del codón 6 del gen de la globina HBB, con un cambio GAG > GTG que causa la sustitución de ácido glutámico por valina (Glu6Val). La ECF es una de las enfermedades genéticas más frecuentes en Estados Unidos, donde se da en 1/2.400 nacimientos. En afroamericanos, la ECF afecta a aproximadamente 1/400 nacimientos y se estima que hay 100.000 individuos con ECF en Estados Unidos60.
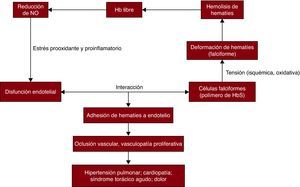
De las múltiples enfermedades cardiovasculares asociadas a esta enfermedad, se ha considerado a la interacción entre hematíes falciformes y endotelio uno de los posibles mecanismos principales de inicio. La ECF es un prototipo de trastorno en que el eritrocito sufre una tensión isquémica, oxidativa o de fuerza tangencial que causa un cambio en su morfología que lo predispone a la polimerización y la consiguiente deformación falciforme. Este cambio causa un aumento de la adhesión entre el eritrocito y la célula endotelial. La disfunción endotelial se caracteriza por reducción de la biodisponibilidad de óxido nítrico (NO), estrés prooxidante y proinflamatorio y coagulopatía que dan lugar a inestabilidad vasomotora y producen finalmente una vasculopatía proliferativa. La lesión endotelial y la inflamación contribuyen de manera importante a la fisiopatología de la ECF y a los síndromes de talasemia β (Figura 1)61.

Figura 1. Afecciones cardiovasculares de la enfermedad de células falciformes. Las células falciformes con polímeros de hemoglobina S sufren una tensión isquémica, oxidativa o de fuerza tangencial. Ello causa deformaciones de los hematíes, denominadas «falciformes», y la consiguiente hemolisis. La hemoglobina libre, liberada al plasma por la hemólisis, elimina el óxido nítrico y da lugar a disminución de la biodisponibilidad de este. La disfunción endotelial caracterizada por la reducción de la biodisponibilidad de óxido nítrico, el estrés prooxidante y proinflamatorio y la coagulopatía da lugar a un aumento de la adhesión de los eritrocitos a las células endoteliales, causa oclusión vascular y vasculopatía proliferativa y finalmente conduce a una serie de manifestaciones, como dolor, hipertensión pulmonar, síndrome pulmonar agudo, ictus e infarto de miocardio. Hb: hemoglobina; HbS: hemoglobina S; NO: óxido nítrico.
La manifestación más frecuente de la ECF es la crisis vasooclusiva, caracterizada por episodios de dolor inesperados e intermitentes. El estrés hemodinámico causado al corazón puede manifestarse en forma de cardiomegalia e isquemia miocárdica62. La hipertensión pulmonar es otra consecuencia conocida de la anemia de células falciformes. Se produce en un 30-40% de los pacientes con ECF y se asocia a un aumento de la mortalidad63. Los trastornos premórbidos más frecuentes en pacientes con ECF son: síndrome torácico agudo/neumonía (58,1%), hipertensión pulmonar (41,9%), hipertensión sistémica (25,6%), insuficiencia cardiaca congestiva (25,6%), infarto de miocardio (20,9%) y arritmias (14,0%)64.
Las células falciformes anormales en sangre identificadas al microscopio indican la existencia de una ECF. El examen se realiza habitualmente en un frotis de sangre, utilizando una preparación especial pobre en oxígeno. Pueden usarse también otras preparaciones para detectar la HbS anormal, como las pruebas de solubilidad realizadas en tubos de soluciones de sangre. La enfermedad puede confirmarse cuantificando específicamente los tipos de hemoglobina presentes mediante electroforesis de la hemoglobina.
La mayor parte de la evidencia actualmente existente indica que la hidroxiurea es bien tolerada, segura y eficaz en la mayoría de los pacientes con ECF65. Para las complicaciones sería útil disponer de nuevos agentes terapéuticos dirigidos a la vía del NO. Los resultados de los estudios en los que se ha examinado el efecto de NO inhalado en diferentes subtipos de ECF parecen ser prometedores, a pesar del estado de resistencia al NO que se observa en los pacientes y en modelos animales66. Se ha demostrado que el sildenafilo, un inhibidor de la fosfodiesterasa tipo 5, mejora la hipertensión pulmonar en los pacientes con ECF67. Serán necesarios ensayos clínicos amplios para confirmar su eficacia y su seguridad en la población de células falciformes.
ConclusionesEn las últimas dos décadas se han producido avances importantes en nuestro conocimiento de los trastornos hematológicos, gracias al estímulo del rápido crecimiento de la biología molecular, la genética y las plataformas diagnósticas contemporáneas. Hemos presentado una revisión centrada en los trastornos hematológicos no oncológicos que afectan a las proteínas de la coagulación del plasma, las plaquetas y los hematíes, y sus posibles repercusiones en el sistema cardiovascular, incluidos los fenotipos frecuentes de infarto de miocardio, ictus isquémico y episodios oclusivos arteriales periféricos. Se ha resaltado también la TEV, tomándola como patrón de referencia clínico para aumentar el conocimiento de un problema frecuente al que se enfrentan todos los clínicos, incluidos los cardiólogos generales, y para diferenciar claramente los trastornos sanguíneos que son específicos del sistema circulatorio venoso por oposición al arterial. Por último, se han presentado pasos prácticos y una guía general para las pruebas diagnósticas y el manejo de la asistencia clínica habitual, con el objeto de facilitar una asistencia segura, efectiva y coste-eficiente de los pacientes.
Conflicto de interesesTracy Y. Wang: subvenciones de investigación de Bristol-Myers Squibb/Sanofi Partnership, Schering Plough, The Medicines Co, Heartscape, Canyon Pharmaceuticals, Eli Lilly/Daiichi Sankyo Alliance; Consultor de Medco, Astra Zeneca.
Richard C. Becker: subvenciones de investigación de Astra Zeneca, Bayer Pharmaceuticals, BMS, Daiichi, Eli Lilly, Johnson & Johnson, Merck, Momenta Pharmaceuticals y Regado Biosciences Inc.
Recibido 18 Febrero 2011
Aceptado 22 Febrero 2011
Autor para correspondencia: Divisions of Cardiology and Hematology, Duke University School of Medicine, Duke Clinical Research Institute, 2400 Pratt Street, Durham, NC 27705, Estados Unidos. richard.becker@duke.edu