The fundamental relationship between blood disorders and the cardiovascular system originates within multiple points of interface, ranging from the heart and its structural constituents to include heart chambers, valves, coronary arteries, coronary veins, and the cerebrovascular and peripheral vasculature. While the cellular components of circulating blood derive their primary origin from multipotent progenitor cells, plasma-based components, which include coagulation proteins, are mostly born of hepatic synthesis and endothelial cells. Here we provide a focused overview of nononcological blood disorders and their potential impact on the arterial circulatory system as common phenotypes, including myocardial infarction, ischemic stroke and peripheral arterial occlusive events. Venous thromboembolism is employed in our discussion as a clinical template. We also provide practical steps and guidance for diagnostic testing and management in routine clinical practice.
Keywords
.
INTRODUCTIONBlood is the medium for exchanging oxygen, nutrients, and waste products throughout the body, and consists of plasma, blood cells (red blood cells, white blood cells), and platelets. Platelets play an important role in clotting, white blood cells are responsible for inflammation, and red blood cells carry oxygen and nutrients to all tissues of the body and carry waste products away from the organs. Any abnormality of these blood components can result in hematologic disorders. While disorders involving platelets and coagulation that can lead to thrombosis and/or bleeding are of primary concern for most cardiologists, disorders involving red blood cells and platelets can also affect the mechanics of blood flow and blood viscosity. Our understanding of hematologic disorders has advanced steadily in recent years, particularly with the development of genetics and molecular biologic techniques. Here we provide a focused overview of nononcologic blood disorders and their potential impact on the arterial circulatory system, including the common phenotypes of myocardial infarction, ischemic stroke, and peripheral arterial occlusive events. Venous thromboembolism (VTE) is employed as a clinical template to heighten awareness of a common problem faced by all clinicians and to distinguish blood disorders which are unique to the venous and to the arterial circulatory systems. We also provide practical steps and general guidance for diagnostic testing and management in routine clinical care.
BLOOD DISORDERS OF COAGULATIONVenous thromboembolism is an important and growing public health problem. An estimated 900 000 patients present with clinically evident VTE in the United States each year, resulting in an estimated 300 000 deaths from pulmonary embolism.1 The urgent task for clinical cardiologists is to understand the burden of disease and potential causes of VTE, which has enormous potential to prevent and reduce death and morbidity from VTE.
INHERITED THROMBOPHILIASThe World Health Organization/International Society of Thrombosis and Hemostasis in 1995 defined thrombophilia as an unusual tendency toward thrombosis, which is characterized by features such as early age of onset; recurrent episodes; strong family history; unusual, migratory, or widespread locations; and severity out of proportion to any recognized stimulus. It also refers to hypercoagulable states which are the end result of diseases, disorders, or conditions that heighten one's propensity to form blood clots within the venous, arterial, and/or microcirculatory systems. Primary characteristics for common inherited thrombophilias are summarized in Table 1.
Table 1. Summary of Main Characteristics of Inherited Thrombophilias
| Diseases | Mutations | Prevalence | Manifestation | ||
| VTE | RVTE | AT | |||
| ATD | SERPINC1 gene | 1/500∼1/5000 | + | + | − |
| PCD | Protein C gene | 1/200∼1/500 | + | + | +/− |
| PSD | PROS1 | 0.03%-2% | + | + | +/− |
| APC resistance | FV Leiden (A506G); FVR2 (H1299R); FV Cambridge (R306T) and FV Hong Kong (R306G) | 4%-6% | + | + | − |
| Prothrombin G20210A | Prothrombin G20210A | 2%-4% | + | +/− | − |
−: no evidence; +: the risk increases; APC, activated protein C; AT, arterial thrombosis; ATD, antithrombin deficiency; FV, factor V; PCD, protein C deficiency; PSD, protein S deficiency; RVTE, recurrent venous thromboembolism; VTE, venous thromboembolism.
Antithrombin is a single-chain glycoprotein belonging to the serine protease inhibitor (serpin) super family that plays a key anticoagulant role by preventing inappropriate, excessive, or mislocalized clotting of blood, which may cause thrombotic disorders. To date, up to 228 distinct mutations have been described in the SERPINC1 gene associated with antithrombin deficiency,2 with a reported prevalence of antithrombin deficiency of 1 in 500 to 1 in 5000 in the overall population.3 Antithrombin deficiency is associated with increased risks of pulmonary embolism and upper and lower extremity deep VTE, but VTE can also occur in unusual sites such as cerebral or sinus, mesenteric, portal, hepatic, renal, and retinal veins. A metaanalysis of observational studies reported that antithrombin deficiency significantly increased the risk of first VTE with odds ratio (OR) 8.73 (95% confidence interval [CI] 3.12-24.42), and recurrent VTE with OR 3.37 (95% CI 1.57-7.20).4 Despite its clear relationship with VTE, beginning as early as the first decade of life, there is no clear evidence that antithrombin deficiency increases the risk for arterial thrombosis.5, 6
Protein C DeficiencyProtein C is a Vitamin K-dependent glycoprotein synthesized by the liver. Thrombin complexed to endothelial cell-based thrombomodulin cleaves an arginine-leucine bond within protein C, causing its activation. In turn, activated protein C inactivates factors Va and XIIIa, thus affecting both contact activated and tissue factor mediated pathways of the coagulation pathway. Activated protein C also neutralizes plasminogen activator inhibitor I, thus enhancing fibrinolytic activity. More than 160 protein C gene mutations have been reported,7 and congenital protein C deficiency is transmitted in an autosomal dominant pattern. The prevalence of asymptomatic protein C deficiency has been reported to be between 1 in 200 and 1 in 500 healthy individuals, whereas the prevalence of clinically significant protein C deficiency has been estimated at 1 in 20 000 patients.8
The clinical phenotype of simple heterozygous protein C deficiency can range from asymptomatic to severe VTE. An estimated 50% of carriers develop venous thrombosis by age 45.9 A metaanalysis reported that protein C deficiency significantly increased the risk of first VTE (OR=7.75, 95% CI 4.48-13.38), and recurrent VTE (OR=2.53, 95% CI 1.30-4.92).4 In addition to VTE, the patients with heterozygous protein C deficiency may be at risk for arterial ischemic stroke or mesenteric artery thrombosis. Another systematic review reported a pooled OR of 6.49 (95% CI 2.96-14.27) for arterial ischemic stroke among the patients with protein C deficiency.5 However, there is still no evidence of association between early signs of atherosclerotic alterations (intima-media thickness, ankle/brachial pressure index) and protein C deficiency.6 Considered collectively, protein C deficiency may be a risk factor for arterial ischemic stroke, particularly in patients younger than 55 years.
Protein S DeficiencyProtein S is a Vitamin K-dependent glycoprotein that is 40% unbound and active in the circulation, and acts as the principle co-factor of activated protein C, increasing the protein's affinity for negatively charged phospholipids. The resulting membrane-bound activated protein C-protein S complex produces a marked increase in Factor Va and VIIIa inactivation. Hereditary protein S deficiency is an autosomal dominant disorder with almost 200 different PROS1 mutations resulting in loss of function identified.10, 11
Although protein S deficiency is uncommon in the general population, it is found in approximately 2% of unselected patients and 1%–13% of thrombophilic patients with VTE respectively.12 Individuals with protein C deficiency experience a heightened risk for VTE and recurrent VTE. A metaanalysis reported that protein S deficiency significantly increased the risk of first VTE, with an OR of 5.77 (95% CI 3.07-10.85), and recurrent VTE with an OR of 3.76 (95% CI 1.76-8.04).4 In a large cohort of families with hereditary protein S deficiency, the annual incidences of recurrent VTE were 8.4% (95% CI 5.8-11.7).13 The results from a large family cohort study also showed that subjects with protein S deficiency have a higher risk (hazard ratio [HR] 4.6, 95% CI 1.1-18.3) for arterial thrombosis before age 55 years.14 However, there are also studies reporting no convincing evidence for a link between protein S deficiency and arterial thrombosis.5, 6
Activated Protein C ResistanceActivated protein C exerts anticoagulant effects beyond inactivating factor Va by cleaving at R306 and R506, generating inactive factor V (FVai). In addition, activated protein C generates an anticoagulant molecule (FVac) by cleaving full-length factor V (FV) at R506; this FVac acts as a cofactor with activated protein C to degrade factor VIIIa. The most common mutation in the FV gene is a single point mutation that results in the replacement of Arg506 in one of the activated protein C cleavage sites with a Gln. This FV Leiden mutation accounts for 90% to 95% of all causes of activated protein C resistance15 and is the most prevalent hereditary thrombophilia, occurring in 4% to 6% of the general population.16 It is rare or absent in populations from Far East Asia, black Africans, and indigenous populations of America and Australia.17 Less common FV mutations also affect activated protein C resistance. Of these, FVR2 (H1299R) reduces activated protein C cofactor activity and leads to an increased thrombotic risk when present in compound heterozygotes with FV Leiden.18 FV Cambridge (R306T) and FV Hong Kong (R306G) are rare mutations that exhibit only mild activated protein C resistance and have not been associated with increased risk of thrombosis.19
Heterozygosity for FV Leiden yields a lifelong hypercoagulable state associated with an approximately 3- to 7-fold increased risk of venous thrombosis, and is also predictive of recurrent VTE.4, 16 In addition to pulmonary embolism and deep venous thromboembolism, FV Leiden significantly increases the risk of first cerebral vein thrombosis with OR of FV 3.38 (95% CI 2.27-5.05).20 FV Leiden is considered a modest risk factor for ischemic stroke and myocardial infarction before age 55 years, particularly among women who smoke. It is not considered to be a risk factor for peripheral arterial occlusive events, with the exception of some patients with advanced native disease and/or surgical graft disease.5, 6, 16
Factor II (Prothrombin) G20210A Gene MutationProthrombin G20210A is a gain-of-function mutation located in the 3’ untranslated region of the prothrombin gene (nucleotide 20210 G to A), leading to increased plasma levels of prothrombin and a hypercoagulable state. Prothrombin G20210A is the second most common inherited risk factor for VTE. Its prevalence depends on geographic location and ethnic background. It is found in 2% to 4% of healthy individuals in southern Europe, which is twice as high as that in northern Europe. Like FV Leiden, it is rare in Far East Asian, African, and indigenous Australian and American populations. In the literature, the mutation is found in 6% to 8% of VTE patients.21
Prothrombin G20210A mutation is associated with a 2- to 3-fold increased risk of VTE,4 and is also associated with recurrent VTE.4, 22 As with the FV Leiden gene mutation, homozygosity is associated with the greatest overall risk.23 There has not been a consistent association of prothrombin G20210A with arterial thrombosis; however, a modestly increased risk is likely present among affected individuals younger than 55 years.5
ACQUIRED THROMBOPHILIASThe most common acquired factors which predispose to thrombosis are presence of an antiphospholipid antibody and malignancy. Antiphospholipid syndrome will be reviewed here. Other less common causes include myeloproliferative disorders, nephrotic syndrome, paroxysmal nocturnal hemoglobinuria; and iatrogenic etiologies, such as chemotherapy for cancer, hormonal treatment for infertility, and surgical procedures.
Antiphospholipid SyndromeAntiphospholipid syndrome is an autoimmune disorder characterized by the clinical association of antiphospholipid antibody with a condition of hypercoagulability, and poses high risk for both VTE and arterial thrombosis.24 Antiphospholipid antibody syndrome is associated with myocardial infarction, intracardiac thrombosis, and pulmonary hypertension resulting from the predisposition to thrombosis, and, less frequently, with valvular heart disease and atherosclerosis of peripheral and coronary arteries. The latter might be explained by antibody-mediated proinflammatory and procoagulant effects exerted directly on endothelial cells.25 Antiphospholipid syndrome patients have significantly worse cardiovascular outcomes.26
DIAGNOSTIC STRATEGIES FOR VENOUS AND ARTERIAL THROMBOSISWhen venous or arterial thrombosis occurs and the suspicion of an acquired thrombophilia is raised, screening tests for lupus anticoagulant are usually performed. Current antiphospholipid syndrome diagnostic criteria require a positive test in the antiphospholipid antibodies panel test (lupus anticoagulant, moderate or high titer anticardiolipin antibodies, and/or anti-b2GP1 antibodies) on 2 separate occasions at least 12 weeks apart, in the setting of thrombosis or pregnancy complications.27 A workup for inherited thrombophilia is usually indicated only in patients with a history of multiple thromboembolic episodes, thromboembolism at a young age, family history of thromboembolism, thrombosis in an unusual site, or VTE without any obvious risk factor. The diagnosis of inherited thrombophilias can be approached employing the following steps: a) functional amidolytic assays should be performed in each of the abovementioned inherited thrombophilias except for the prothrombin G20210A gene mutation, which can be tested directly using genetic analysis; b) functional tests are performed to exclude antithrombin deficiency, protein C deficiency, and protein S deficiency; and c) genetic testing is required for the detection of specific gene mutations such as FV Leiden. Functional assays are preferably avoided in the setting of acute thrombosis and best performed before or several days after cessation of heparin and/or oral anticoagulation therapies, as acute thrombosis and anticoagulation therapy may affect the results. However, genetic assays for G20210A or FV Leiden can be performed at any time. All causes of acquired thrombophilias should be excluded before classifying a person with abnormal test results as having an inherited thrombophilia. Testing among patients with arterial thrombosis, including the common phenotypes of myocardial infarction, ischemic stroke and/or peripheral arterial occlusive events should be individualized, with a primary focus on traditional risk factors, systemic diseases associated with atherosclerotic vascular disease, and antiphospholipid syndrome with or without a circulating Lupus anticoagulant prior to embarking on testing for hereditary thrombophilias in patients younger than 55 years.
PREVENTION AND TREATMENT STRATEGIES FOR VENOUS AND ARTERIAL THROMBOSISAsymptomatic individuals: Long-term anticoagulant thromboprophylaxis is not recommended in asymptomatic patients with inherited thrombophilias because of the increased risk of hemorrhage.26
Prophylaxis in high-risk settings: Heparin or low-molecular-weight heparin should be considered strongly for thromboprophylaxis when individuals with inherited thrombophilias and prior arterial thrombosis find themselves in high-risk settings, including major surgery, trauma, or management of pregnancy, labor, and delivery.28
Venous and arterial thrombosis treatment: The initial management of coronary arterial thrombosis in patients with an inherited thrombophilia should proceed according to the standard of care, with anticoagulant and platelet-directed therapy as indicated. Consideration of long-term anticoagulant therapy must be individualized in the absence of randomized studies. The management of VTE is usually not different from that of VTE in other patients without inherited thrombophilias.28
The cornerstone of management for acquired thrombophilias is treatment of the underlying disease. Patients who are persistently positive for antiphospholipid antibodies, and who have a documented history of either VTE or arterial thromboembolism, are at increased risk of recurrence. Long-term oral anticoagulant therapy is the mainstay of treatment, with a target international normalized ratio of 2.0 to 3.0. There is no general consensus on the prophylactic treatment of antiphospholipid antibody carriers who have not experienced vascular/thrombotic or obstetric manifestations.29 Acetylsalicylic acid prophylaxis may be sufficient in low-risk settings.
DISORDERS OF PLATELETS Heparin-induced ThrombocytopeniaHeparin-induced thrombocytopenia (HIT), an immune reaction in response to platelet factor 4-heparin complexes, can occur in 0.1% to 5% of patients receiving heparin, depending on the patient population, source and formulation of heparin, and dose and duration of treatment. HIT occurs more frequently in surgical patients than in medical patients, and more often with unfractionated heparin than with low molecular weight heparin.30 HIT is characterized by thrombocytopenia and a strong propensity for paradoxical thrombosis, manifesting either arterial, microvascular, or VTE, the latter being more common.31, 32
HIT is diagnosed using a combination of clinical and laboratory criteria. Two principal criteria are essential for establishing a clinical diagnosis: development of thrombocytopenia and/or clinical thrombosis in temporal association with heparin therapy (typically within 5 to 14 days of heparin exposure),33 and exclusion of other causes of thrombocytopenia. Detection of HIT antibodies is necessary, but not sufficient, for the diagnosis because only a subset of individuals who develop heparin antibodies actually develop HIT. There are several available assays to detect HIT antibodies, including the serotonin release assay (SRA), enzyme-linked immunosorbant assay (ELISA), or enzyme immunoassay (EIA).
Heparin cessation and initiation of an alternative anticoagulant should occur immediately after clinical suspicion is aroused and even before the result of any serologic test becomes available.34 Alternative anticoagulants include the direct thrombin inhibitors lepirudin, argatroban, and bivalirudin, and the anti-Xa agent fondaparinux.34 The use of warfarin anticoagulation has been associated with the development of venous limb gangrene and should be avoided until the platelet count has improved. Furthermore, it should be initiated with concurrent nonheparin anticoagulation and without bolus dosing.34 For patients with HIT who have not sustained a thromboembolic event, therapeutic anticoagulation should be continued until the platelet count has returned to a stable plateau.34 HIT patients with a thromboembolic complication should receive a standard course of therapeutic anticoagulation for the specific clinical event.34 The American College of Chest Physicians consensus statement recommends that platelet counts should be monitored every 2 to 3 days beginning on the 4th day after initiation of therapy, until the therapy is discontinued or until the 14th day of heparin exposure.34
Thrombotic Thrombocytopenic PurpuraThrombotic thrombocytopenic purpura (TTP) is a life threatening multisystem disorder caused by platelet and von Willebrand factor deposition in arterioles and capillaries which, in turn, prompts widespread organ ischemia, particularly affecting the brain, heart, and kidneys. Microangiopathic hemolytic anemia ensues as the red blood cells pass through the affected vessels and break into fragments called schistocytes.35 A rare condition resulting from the deficiency of ADAMTS13, TTP affects approximately 1000 persons per year in the United States, is much more prevalent among women, and the incidence among African Americans is 9 times higher than in other ethnicities.36 The classic pentad of thrombocytopenia, hemolytic anemia, fever, renal dysfunction, and neurologic symptoms is present in only a minority of TTP patients.37 A single-center study showed that 21% of TTP patients develop incident myocardial infarction.38 Among cardiovascular patients, use of antiplatelet agents such as clopidogrel and ticlopidine has been rarely associated with drug-induced TTP. Clopidogrel-associated TTP often occurs within 2 weeks of drug initiation. The survival rate for patients with clopidogrel-associated TTP is approximately 71.2%.39
The clinical laboratory picture is similar to that of disseminated intravascular coagulation, although prothrombin time and activated partial thromboplastin time are rarely abnormal.40 ADAMTS-13 activity assays have advanced the diagnosis of TTP, with sensitivities ranging from 89% to 100% and specificity greater than 91%.41 Plasma exchange or infusion is the mainstay of treatment for TTP. Glucocorticoids, cyclosporine, vincristine, splenectomy, and, more recently, rituximab (monoclonal antibody to CD20) have all been used in combination with plasma exchange to treat TTP, although randomized clinical trial data are lacking.42 On the other hand, some have suggested a role for the therapeutic use of antiplatelet agents in conjunction with plasma exchange despite significant thrombocytopenia.42 Similarly, a previous standard of care held that platelet transfusion was contraindicated in TTP, but a recently published review concludes that it is still uncertain whether this practice is harmful or not.43
Immune ThrombocytopeniaImmune thrombocytopenia (ITP) is an acquired immune-mediated disorder characterized by isolated thrombocytopenia (platelet count 150×109/L), in the absence of other causes of thrombocytopenia. Although the development of autoantibodies against platelet glycoproteins remains central in the pathophysiology of ITP, several abnormalities involving the cellular mechanisms of immune modulation have been identified.44 It appears both platelet survival and production are impaired in ITP.
An analysis from the General Practice Research Database in the United Kingdom showed that the current estimate of the incidence of ITP is 3.3 per 105 adults/year for adults.45 Feudjo-Tepie et al. reported the prevalence of ITP in the years 2002–2006 for adults and the overall population was 23.6 and 20.3, respectively, per 100 000 in the United States.46 The symptoms and signs are highly variable and range from the completely asymptomatic patient to frank hemorrhage from any site, the most serious of which is intracranial. Even if the platelet count is low, myocardial infarction and ischemic stroke can occur in some patients.47
The diagnosis of ITP is made by excluding other causes of thrombocytopenia. The basic diagnostic approach to ITP includes a patient history, physical examination, complete blood count, detection of antiplatelet antibodies and examination of a peripheral blood smear. Bone marrow aspiration in patients older than 60 years is appropriate to rule out leukemia, infiltrative disease and aplastic anemias.
Treatment of patients with ITP must take into account the age of the patient, the severity of the illness, and the anticipated natural history. Adult patients, particularly those older than 60 years, have a higher incidence of major or fatal bleeding than children. However, specific therapy may not be necessary unless the platelet count is <20×109/L or there is extensive bleeding. In fact, the current treatment for ITP is considered appropriate for symptomatic patients and for those at risk of bleeding.48 Provided the patient's situation is not life threatening, corticosteroids are the standard initial treatment for the ITP. Intravenous immune globulin is generally recommended for patients unresponsive to corticosteroids. The platelet count also can be supported by anti-D immunoglobulin, which is active only in Rh-positive patients and in the pre-splenectomy setting.48 Splenectomy is traditionally considered to be the second-line treatment in adults with ITP in whom achieving a safe platelet count with initial prednisone therapy has failed, and it is effective for most patients. The treatment of chronic ITP has advanced in recent years. These advances include the incorporation of immunomodulatory therapy (rituximab, anti-CD20 monoclonal antibody) and the development of thrombopoietic stimulating agents (romiplostim, eltrombopag), which has been used in clinical trials and showed some good benefits,49 but any long-term adverse impact is unknown.
Following percutaneous coronary interventions, patients with ITP have risk for bleeding or thrombotic complications when antiplatelet treatment is given or spared, respectively. Given the paucity of data on ITP and stenting, no strict recommendations can be proposed and treatment should be individualized to minimize both bleeding and thrombosis risks. Nonetheless, several cases suggest the feasibility of percutaneous revascularization in selected patients with multivessel coronary disease and ITP.47
ABNORMALITIES OF RED BLOOD CELLS AnemiaThe World Health Organization defines anemia as a hemoglobin concentration <13 g/dL in men and <12 g/dL in postmenopausal women. Anemia is common among patients with cardiovascular disease. The prevalence of anemia in patients with congestive heart failure ranges between 4% and 61% (median 18%) depending on the studied population, with the majority of studies indicating prevalence >20%.50 Anemia appears to be more prevalent in patients with advanced age and in those with severe limitations in functional capacity and greater severity of co-morbid kidney disease.50 Among patients with myocardial infarction, baseline anemia was present in 10.5% of patients51 and the incidence of hospital-acquired anemia was high (range 33% to 69% depending on hospital).52
The etiologies of anemia are multifactorial, including blood loss in the context of antiplatelet or anticoagulant therapies, iron deficiencies, hemodilution, activation of the inflammatory cascade, urinary losses of erythropoietin, and associated renal insufficiency.52, 53 Anemia is a strong independent predictor of increased mortality, hospitalization, and bleeding risk in patients with cardiovascular disease.51, 52, 53
Treatment and management of anemia has centered on erythropoietin-stimulating agents and parenteral iron supplementation. Many studies show a positive effect of erythropoietin or its derivatives when administered in combination with oral or intravenous iron, showing improvements in left and right ventricular systolic and diastolic function, dilation and hypertrophy, and renal function.54 There is a need for an adequately powered randomized phase III trial. The STAMINA-HeFT study, a double-blind, randomized, placebo-controlled, multicenter trial, showed a nonsignificant trend toward a lower risk of all-cause mortality or first heart failure hospitalization in darbepoetin-alfa-treated patients, compared with placebo (HR 0.68; 95% CI 0.43, 1.08; P=.10).55 However, the Trial to Reduce Cardiovascular Events with Aranesp Therapy (TREAT) reported that the use of darbepoetin-alfa in patients with diabetes, chronic kidney disease, and moderate anemia who were not undergoing dialysis did not reduce the risk of either of the primary composite outcomes (ie, death or a cardiovascular event and death or a renal event) and was associated with an increased risk of stroke.56 Accordingly, the optimal threshold at which therapy should be initiated and the extent of correction considered safe and desirable in the individual patient with heart failure have not been established. The second mortality and morbidity trial, Reduction of Events with Darbepoetin alfa in Heart Failure (RED-HF), is in progress and is likely to provide more answers.57 More recently, the Reduction of Infarct Expansion and Ventricular Remodeling With Erythropoietin After Large Myocardial Infarction (REVEAL) trial led by Rao et al., a randomized, double-blind, placebo-controlled, multicenter trial, is evaluating the effects of epoetin α on infarct size and left ventricular remodeling in patients with large myocardial infarctions. Its results, presented at the American Heart Association 2010 Scientific Sessions in Chicago, show that an intravenous injection of erythropoietin following successful primary or rescue percutaneous coronary intervention did not reduce infarct size in ST-segment-elevation myocardial infarction patients.58 The available evidences show that the routine use of either transfusions or bone marrow stimulating agents do not benefit, and may in fact do harm to, patients with myocardial infarction or heart failure, even those with concomitant renal insufficiency.59
Sickle Cell DiseaseSickle cell disease (SCD) is an inherited genetic disorder characterized by a hemoglobin abnormality called “hemoglobin S” (HbS). It refers to a group of hemolytic anemias in which HbS is present in either a homozygous state (HbSS) or a compound heterozygous state, such as when combined with hemoglobin C (HbSC) or β-thalassemia (HbS–β-thalassemia). SCD is caused by homozygosity for a single nucleotide mutation in codon 6 of the HBB globin gene, GAG>GTG, resulting in the substitution of valine for glutamic acid (Glu6Val). SCD is one of the most common genetic diseases in the United States, occurring in 1 in 2400 births. Among African Americans, SCD affects approximately 1 in 400 births and it is estimated that there are 100 000 individuals in the United States with SCD.60
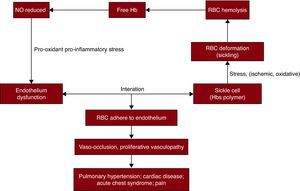
Among the multiple cardiovascular pathologies associated with this disease, a sickle red cell-endothelial interaction has been implicated as one of the major potential initiating mechanisms. SCD is a prototype of a condition in which the erythrocyte is under ischemic, oxidative, or shear stress that results in changes in the erythrocyte morphology, predisposing to polymerization and consequent deformation (“sickling”). This change leads to enhanced erythrocyte-endothelial cell adhesion. The endothelial dysfunction is characterized by reduced nitric oxide (NO) bioavailability, pro-oxidant and pro-inflammatory stress and coagulopathy, leading to vasomotor instability and ultimately producing a proliferative vasculopathy. Endothelial damage and inflammation make a significant contribution to the pathophysiology of SCD and the β-thalassemia syndromes61 (Figure 1).

Figure 1. Cardiovascular pathologies of sickle cell disease. The sickle cells with hemoglobin S polymers are under ischemic, oxidative, or shear stress. These conditions cause deformations to red blood cells, called sickling and hemolysis. Free hemoglobin,released into plasma by hemolysis scavenges on nitric oxide, and leads to reduced nitric oxide bioavailability. The endothelial dysfunction characterized by reduced nitric oxide bioavailability, pro-oxidant and pro-inflammatory stress, and coagulopathy leads to enhanced erythrocyte-endothelial cell adhesion, results in vaso-occlusion and proliferative vascularopathy, and eventually produces a series of manifestations, such as pain, pulmonary hypertension, acute chest syndrome, stroke, and myocardial infarction.Hb: hemoglobin; HbS: hemoglobin S; NO: nitric oxide; RBC: red blood cells.
The most common manifestation of SCD is vaso-occlusive crisis, characterized by intermittent, unexpected episodes of pain. Hemodynamic stressors to the heart can present as cardiomegaly and myocardial ischemia.62 Pulmonary hypertension is another known consequence of sickle cell anemia. It occurs in 30% to 40% of patients with SCD, and is associated with increased mortality.63 The most common premorbid conditions among patients with SCD include acute chest syndrome/pneumonia (58.1%), pulmonary hypertension (41.9%), systemic hypertension (25.6%), congestive heart failure (25.6%), myocardial infarction (20.9%), and arrhythmias (14.0%).64
SCD is suggested when the abnormal sickle-shaped cells in the blood are identified under a microscope. Testing is typically performed on a smear of blood using a special low-oxygen preparation. Other prep tests can also be used to detect the abnormal HbS, including solubility tests performed on tubes of blood solutions. The disease can be confirmed by specifically quantifying the types of hemoglobin present using a hemoglobin electrophoresis test.
The bulk of the current evidence suggests that hydroxyurea is well tolerated, safe, and efficacious for most patients with SCD.65 Novel therapeutic agents focusing on the NO pathway would be beneficial for complications. Results from studies that examined the effect of inhaled NO on different SCD subtypes appear to be promising despite the state of NO resistance observed in patients and in animal models.66 Sildenafil, a phosphodiesterase type 5 inhibitor, has been shown to improve pulmonary hypertension in SCD patients.67 Large trials are needed to confirm their efficacy and safety in the sickle cell population.
CONCLUSIONSOur understanding of hematologic disorders has advanced steadily over the past two decades, stimulated by rapid growth in molecular biology, genetics, and contemporary diagnostic platforms. We have provided a focused overview of nononcological blood disorders involving plasma coagulation proteins, platelets, and red blood cells and their potential impact on the cardiovascular system, including the common phenotypes of myocardial infarction, ischemic stroke, and peripheral arterial occlusive events. Venous thromboembolism was highlighted as well, serving as a clinical template to heighten awareness of a common problem faced by all clinicians, including general cardiologists, and to clearly distinguish blood disorders which are unique to the venous as compared to the arterial circulatory systems. Finally, practical steps and general guidance for diagnostic testing and management in routine clinical care were offered to foster safe, effective, and cost-efficient patient care.
CONFLICTS OF INTERESTTracy Y. Wang: Research grants from Bristol-Myers Squibb/Sanofi Partnership, Schering Plough, The Medicines Co, Heartscape, Canyon Pharmaceuticals, Eli Lilly/Daiichi Sankyo Alliance; Consultant for Medco, Astra Zeneca. Richard C. Becker: Research grants from Astra Zeneca, Bayer Pharmaceuticals, BMS, Daiichi, Eli Lilly, Johnson & Johnson, Merck, Momenta Pharmaceuticals, and Regado Biosciences Inc.
Received 18 February 2011
Accepted 22 February 2011
Corresponding author: Divisions of Cardiology and Hematology, Duke University School of Medicine, Duke Clinical Research Institute, 2400 Pratt Street, Durham, NC 27705, United States. richard.becker@duke.edu