La amiloidosis hereditaria por transtiretina es una enfermedad autosómica dominante causada por mutaciones en el gen de la transtiretina. Se han descrito hasta la fecha más de 100 mutaciones, de las que Val30Met es la más común; en los casos con daño neurológico predominante se denomina polineuropatía amiloidótica familiar (PAF) o enfermedad de Corino-Andrade1. La identificación de pacientes cuya amiloidosis se debe a un defecto genético tiene gran importancia, ya que modifica el tratamiento y tiene gran trascendencia para los familiares2. Aunque es muy poco frecuente en todo el mundo, hay descripciones de algunos focos endémicos, y actualmente la isla de Mallorca es el quinto foco mundial en número de afectados, por detrás de países como Portugal, Suecia, Japón y Brasil. Existe asimismo otro foco endémico en España, más reducido, en Valverde del Camino (Huelva)3. La mutación Val30Met se presenta generalmente con neuropatía sensorial periférica y avanza a neuropatía autonómica y motora, con aparición tardía de trastornos de la conducción cardiaca y sin hipertrofia cardiaca, según los datos de la literatura4. Los tratamientos existentes aprobados para la PAF incluyen el trasplante hepático y el tafamidis, un fármaco estabilizante de la transtiretina. Están en estudio otros fármacos, con resultados iniciales prometedores. Actualmente se recomienda iniciar el tratamiento farmacológico o indicar el trasplante hepático cuando se identifiquen los primeros signos y síntomas neurológicos. La amiloidosis cardiaca es una de las principales causas de muerte en la PAF, y en muchos de estos pacientes con daño cardiaco está infradiagnosticada5.
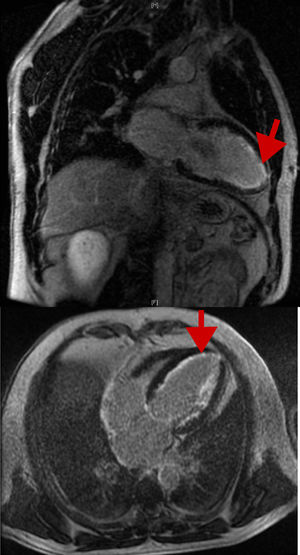
Nuestro objetivo es evaluar el daño cardiaco en una serie amplia de pacientes con PAF, debido a que no está bien caracterizada en la literatura por su baja prevalencia, y mucho menos en España, donde no se han publicado datos. Se revisaron las historias clínicas de pacientes con PAF (estudio genético positivo para mutación Val30Met en el gen de la transtiretina en todos, y presencia de amiloide en biopsia de grasa subcutánea, rectal o glándula salival en los sintomáticos). Se recogieron datos demográficos, clínica, electrocardiografía, ecocardiografía y Holter, así como cardiorresonancia magnética y gammagrafía con difosfonatos si se habían realizado. El daño cardiaco se definió por la presencia de signos o síntomas específicos, arritmias, trastornos de la conducción auriculoventricular (AV), hipertrofia ventricular izquierda en el ECG o el ecocardiograma, o realce tardío en cardiorresonancia magnética. Se analizaron los datos de 132 pacientes (tabla): 104 portadores sintomáticos (78,8%) y 28 portadores asintomáticos (21,2%). La media de edad era 47,4±17 años al diagnóstico y 57,2±16,4 años en el seguimiento; 69 (52,2%) eran varones. De los portadores sintomáticos, 83 (79,8%) tenían polineuropatía y 56 (53,8%), algún síntoma cardiovascular o daño cardiaco: el 15%, palpitaciones; el 10,4%, disnea; el 4%, síncope; el 20,6%, síntomas de disautonomía, y el 9,8%, insuficiencia cardiaca. El ECG fue patológico en 39 (36%), por la presencia de signos de hipertrofia ventricular izquierda, arritmias o alteraciones de la conducción AV: 13 (9,8%) con disfunción sinusal o fibrilación auricular (síndrome bradicardia-taquicardia), 17 (12,9%) con bloqueo AV en diferentes grados y 12 (9,1%) con bloqueo de rama del Haz de His. La media del grosor máximo de la pared del ventrículo izquierdo fue 11,2±3,2mm, con una fracción de eyección del ventrículo izquierdo del 61±6%; 19 pacientes (23,5%) tenían un grosor del ventrículo izquierdo ≥ 11mm y 10 (12,3%), ≥ 15mm. De estos 10, en 5 se realizó cardiorresonancia magnética, y en 3 de ellos se observó realce tardío de gadolinio en localización subendocárdica, en el característico anillo (figura). El 46% tenía disfunción diastólica y 9 (6,8%) precisaron implante de marcapasos durante el seguimiento por disfunción sinusal o bloqueo AV avanzado. A 54 (41%) se les realizó trasplante hepático, 1 paciente con trasplante combinado corazón-hígado, y en los últimos años 11 han recibido tafamidis. Durante el seguimiento 22 (16,7%) fallecieron. Aunque el daño cardiaco fue clínicamente evidente solo en la fase tardía de la enfermedad (los pacientes eran unos 10 años mayores que los que solo tienen fenotipo neurológico), se asoció con mayor comorbilidad y se relacionó estadísticamente con un aumento de la mortalidad (p=0,003). En el análisis multivariable los parámetros relacionados de manera independiente con la mortalidad (p<0,05) durante el seguimiento fueron un ECG patológico, bloqueo AV, insuficiencia cardiaca, marcapasos, afección neurológica e insuficiencia renal. La supervivencia media desde el inicio de los síntomas hasta la muerte fue de 7,3 años. En conclusión, nuestros resultados, basados en una serie grande dada la baja prevalencia de la enfermedad, proporcionan información adicional sobre la PAF y son útiles para la descripción de sus características demográficas, su presentación clínica, su diagnóstico y su evolución. La PAF es una enfermedad rara que no solo genera síntomas neurológicos clásicos, como se había pensado hasta hace unos años, sino que también la afección cardiovascular es frecuente, especialmente ECG patológico, alteraciones del ritmo y trastornos de conducción AV, hipertrofia ventricular izquierda con disfunción diastólica y síntomas de disautonomía, y se relaciona con una mayor morbimortalidad. En el área endémica de Mallorca, en comparación con Portugal, Suecia y Japón, la presencia de hipertrofia ventricular izquierda e insuficiencia cardiaca es más frecuente y confiere un peor pronóstico. El cardiólogo debe realizar un seguimiento estrecho de estos pacientes— con ECG, ecocardiograma y Holter anuales— y ampliar el estudio (cardiorresonancia magnética, gammagrafía con difosfonatos, etc.) en caso de síntomas o alteraciones.
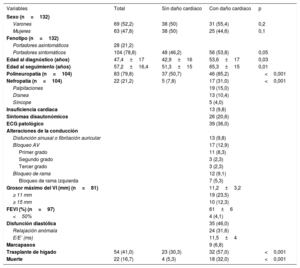
Datos clínicos de los pacientes incluidos en el estudio
| Variables | Total | Sin daño cardiaco | Con daño cardiaco | p |
|---|---|---|---|---|
| Sexo (n=132) | ||||
| Varones | 69 (52,2) | 38 (50) | 31 (55,4) | 0,2 |
| Mujeres | 63 (47,8) | 38 (50) | 25 (44,6) | 0,1 |
| Fenotipo (n=132) | ||||
| Portadores asintomáticos | 28 (21,2) | |||
| Portadores sintomáticos | 104 (78,8) | 48 (46,2) | 56 (53,8) | 0,05 |
| Edad al diagnóstico (años) | 47,4±17 | 42,9±16 | 53,6±17 | 0,03 |
| Edad al seguimiento (años) | 57,2±16,4 | 51,3±15 | 65,3±15 | 0,01 |
| Polineuropatía (n=104) | 83 (79,8) | 37 (50,7) | 46 (85,2) | <0,001 |
| Nefropatía (n=104) | 22 (21,2) | 5 (7,8) | 17 (31,0) | <0,001 |
| Palpitaciones | 19 (15,0) | |||
| Disnea | 13 (10,4) | |||
| Síncope | 5 (4,0) | |||
| Insuficiencia cardiaca | 13 (9,8) | |||
| Síntomas disautonómicos | 26 (20,6) | |||
| ECG patológico | 39 (36,0) | |||
| Alteraciones de la conducción | ||||
| Disfunción sinusal o fibrilación auricular | 13 (9,8) | |||
| Bloqueo AV | 17 (12,9) | |||
| Primer grado | 11 (8,3) | |||
| Segundo grado | 3 (2,3) | |||
| Tercer grado | 3 (2,3) | |||
| Bloqueo de rama | 12 (9,1) | |||
| Bloqueo de rama izquierda | 7 (5,3) | |||
| Grosor máximo del VI (mm) (n=81) | 11,2±3,2 | |||
| ≥ 11 mm | 19 (23,5) | |||
| ≥ 15 mm | 10 (12,3) | |||
| FEVI (%) (n=97) | 61±6 | |||
| <50% | 4 (4,1) | |||
| Disfunción diastólica | 35 (46,0) | |||
| Relajación anómala | 24 (31,6) | |||
| E/E’ (ms) | 11,5±4 | |||
| Marcapasos | 9 (6,8) | |||
| Trasplante de hígado | 54 (41,0) | 23 (30,3) | 32 (57,0) | <0,001 |
| Muerte | 22 (16,7) | 4 (5,3) | 18 (32,0) | <0,001 |
AV: auriculoventricular; ECG: electrocardiograma; FEVI: fracción de eyección del VI; VI: ventrículo izquierdo.
.")
CIBEROBN (CB12/03/30038), Madrid, España.