
La amiloidosis hereditaria por transtirretina (ATTRv) es una enfermedad causada por mutaciones en el gen de la transtirretina que frecuentemente presenta afección cardiaca debido al depósito de amiloide en el miocardio. Nuestro objetivo es describir esta afección en una cohorte española.
MétodosEstudio retrospectivo multicéntrico de pacientes con ATTRv y afección cardiaca provenientes de centros españoles. Se recogieron datos demográficos, clínicos y genéticos.
ResultadosEn 26 centros se incluyó a 181 pacientes, el 65,2% varones, con una mediana de edad al diagnóstico de 62 años. Las mutaciones más frecuentes fueron Val50Met (67,7%) y Val142Ile (12,4%). El principal motivo de consulta fue extracardiaco (69%), principalmente neurológico. La media de la fracción aminoterminal del propéptido natriurético cerebral (NT-proBNP) fue 2.145±3.586 pg/ml. Lo más característico del electrocardiograma fueron el patrón de seudoinfarto (25,9%) y el bloqueo auriculoventricular (25,3%). El grosor ventricular medio fue 15,4±4,1mm. El strain longitudinal estaba reducido en segmentos basales en el 29,4%. Se observó realce tardío subendocárdico difuso en el 58,8%. En la gammagrafía había captación de grados 2-3 en un 75%. En el seguimiento, el 24,9% ingresó por insuficiencia cardiaca, el 34,3% precisó marcapasos y el 31,6%, trasplante hepático. El 32,5% falleció, principalmente por insuficiencia cardiaca (28,8%). Las mutaciones diferentes de Val50Met se asociaron en general con un peor pronóstico.
ConclusionesLa ATTRv cardiaca en España tiene un espectro genético y de afección heterogéneo. El pronóstico es malo principalmente por las complicaciones cardiacas, por lo que son esenciales un diagnóstico y un tratamiento precoces.
Palabras clave
La amiloidosis cardiaca (AC) es una enfermedad de depósito por acumulación extracelular de fibras anómalas en el corazón1. Tradicionalmente se había considerado la forma por cadenas ligeras (AL) como la más frecuente, pero el estudio de la secundaria a transtirretina anómala, ya sea en su forma hereditaria por mutaciones en TTR (ATTRv) o en su forma natural o wild-type (ATTRwt), se ha incrementado en los últimos años gracias a un mejor conocimiento y a los avances en las técnicas de imagen, lo que ha llevado a considerar que la forma ATTRwt es mucho más prevalente que la AL2,3.
La ATTRv es una enfermedad autosómica dominante causada por mutaciones en el gen de la transtirretina. Más de 120 mutaciones se han descrito hasta la fecha, y la mutación Val50Met es la más común. La identificación de pacientes cuya amiloidosis se debe a un defecto genético tiene gran importancia, ya que modifica el tratamiento y tiene gran trascendencia para los familiares, por el tipo de herencia. Aunque es muy poco frecuente en el mundo (tiene una prevalencia estimada<1/100.000 hab.), hay descripciones de algunos focos endémicos, como Portugal, Suecia, Brasil, Japón y 2 regiones españolas: Mallorca y Huelva. En Portugal, primer foco mundial, la prevalencia estimada es de 1/538 hab., mientras que en Mallorca está en torno a 1/3.500, por lo que se la considera el quinto foco mundial4.
La AC es una de las principales causas de morbimortalidad en la ATTRv5. Los tratamientos aprobados para la ATTRv incluyen el trasplante hepático, el tafamidis, el patisirán y el inotersén. Algunos ensayos clínicos ya han demostrado el beneficio de estos fármacos en el tratamiento de la AC6–8.
En España hay pocos datos publicados sobre ATTRv con afección cardiaca5,9. Es importante realizar un diagnóstico precoz de esta enfermedad para identificar a los pacientes que se puedan beneficiar de los nuevos tratamientos.
El objetivo de nuestro estudio es describir las características demográficas, clínicas y genéticas de los pacientes con ATTRv y afección cardiaca en España, cubriendo todo el territorio nacional (tanto los focos endémicos como las zonas no endémicas).
MÉTODOSSe trata de un estudio descriptivo, retrospectivo y multicéntrico de pacientes diagnosticados de ATTRv con afección cardiaca. Se contactó con la gran mayoría de las unidades de insuficiencia cardiaca y unidades de cardiopatías familiares del territorio español para solicitar su participación en el estudio. El estudio fue aprobado por el Comité de ética autonómico de las Islas Baleares. Se obtuvieron los consentimientos informados de todos los pacientes incluidos.
Los pacientes debían disponer de estudio genético positivo para alguna mutación en el gen de la transtirretina. La AC se definió por la presencia de signos típicos en el ecocardiograma o cardiorresonancia+confirmación histológica (presencia de amiloide en biopsia de grasa subcutánea, rectal, de glándula salival, endomiocárdica u otras) o gammagrafía con captación grado 2 o 310. En las zonas endémicas de la mutación Val50Met, bastaba la presencia de la mutación+clínica compatible+hallazgos típicos en ecocardiograma o cardiorresonancia (≥ 13mm en ausencia de hipertensión o valvulopatía aórtica), por la baja sensibilidad de la gammagrafía en este subgrupo particular11.
En cada centro participante se revisaron las historias clínicas de pacientes con ATTRv y afección cardiaca. Se recogieron datos demográficos generales y de comorbilidades, clínica, genética, biomarcadores, electrocardiograma, ecocardiografía, Holter, cardiorresonancia, gammagrafía miocárdica con bisfosfonatos, biopsia, tratamientos, complicaciones y evolución.
Análisis estadísticoSe realizó un estudio descriptivo mediante el cálculo de frecuencias de las variables cualitativas, así como la mediana [intervalo intercuartílico] para las variables cuantitativas. Para estudiar el ajuste a la normalidad se utilizó el test de Kolmogorov-Smirnov. Para las variables categóricas se calcularon porcentajes. Las variables cuantitativas normales se compararon usando el test no paramétrico de la U de Mann-Whitney y las variables cualitativas, mediante el test de la χ2; de aquellas en que la diferencia era estadísticamente significativa, se calculó el intervalo de confianza del 95% (IC95%). En el caso de las mutaciones más prevalentes (Val50Met y Val142Ile), se llevó a cabo un análisis comparativo entre grupos. Se realizó un análisis de supervivencia desde el inicio del seguimiento mediante el método de Kaplan-Meier, tanto para el total de pacientes como para las mutaciones más prevalentes. En todos los casos se consideraron estadísticamente significativos los valores de p <0,05. Para el análisis estadístico se usó Excel 2007 y SPSS v15.0.
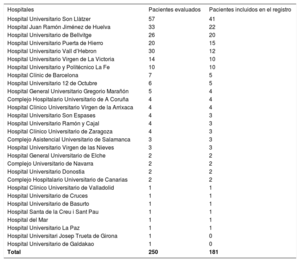
RESULTADOSSe obtuvo información de 250 pacientes y se excluyó a 69 porque no tenían evidencia de afección cardiaca o estaban duplicados (pacientes seguidos o estudiados en 2 centros participantes). El total de hospitales contactados fue 53; de ellos, 27 no tenían ningún caso de ATTRv. Se incluyó finalmente a 181 pacientes de 26 hospitales de España con diagnóstico de ATTRv y afección cardiaca. La distribución por centros se puede consultar en la tabla 1. La mayoría de los pacientes eran varones (65,2%), con una mediana de edad al diagnóstico de 62 años, e inicio de los síntomas a los 58,8±14 años. El tiempo entre el inicio de los síntomas y el diagnóstico de la ATTRv fue de 14 meses. La mediana de seguimiento desde la primera evaluación fue de 34,5 meses. El principal motivo de consulta fue la clínica extracardiaca (38,1%), seguido de la disnea (21,5%) (tabla 2).
Distribución por centros
| Hospitales | Pacientes evaluados | Pacientes incluidos en el registro |
|---|---|---|
| Hospital Universitario Son Llàtzer | 57 | 41 |
| Hospital Juan Ramón Jiménez de Huelva | 33 | 22 |
| Hospital Universitario de Bellvitge | 26 | 20 |
| Hospital Universitario Puerta de Hierro | 20 | 15 |
| Hospital Universitario Vall d’Hebron | 30 | 12 |
| Hospital Universitario Virgen de La Victoria | 14 | 10 |
| Hospital Universitario y Politécnico La Fe | 10 | 10 |
| Hospital Clínic de Barcelona | 7 | 5 |
| Hospital Universitario 12 de Octubre | 6 | 5 |
| Hospital General Universitario Gregorio Marañón | 5 | 4 |
| Complejo Hospitalario Universitario de A Coruña | 4 | 4 |
| Hospital Clínico Universitario Virgen de la Arrixaca | 4 | 4 |
| Hospital Universitario Son Espases | 4 | 3 |
| Hospital Universitario Ramón y Cajal | 4 | 3 |
| Hospital Clínico Universitario de Zaragoza | 4 | 3 |
| Complejo Asistencial Universitario de Salamanca | 3 | 3 |
| Hospital Universitario Virgen de las Nieves | 3 | 3 |
| Hospital General Universitario de Elche | 2 | 2 |
| Complejo Universitario de Navarra | 2 | 2 |
| Hospital Universitario Donostia | 2 | 2 |
| Complejo Hospitalario Universitario de Canarias | 2 | 2 |
| Hospital Clínico Universitario de Valladolid | 1 | 1 |
| Hospital Universitario de Cruces | 1 | 1 |
| Hospital Universitario de Basurto | 1 | 1 |
| Hospital Santa de la Creu i Sant Pau | 1 | 1 |
| Hospital del Mar | 1 | 1 |
| Hospital Universitario La Paz | 1 | 1 |
| Hospital Universitari Josep Trueta de Girona | 1 | 0 |
| Hospital Universitario de Galdakao | 1 | 0 |
| Total | 250 | 181 |
Perfil clínico y genético de los pacientes
| Total | |
|---|---|
| Características basales | |
| Pacientes | 181 |
| Varones | 118 (65,2) |
| Edad al inicio de los síntomas (años) | 60 [51-68] |
| Tiempo síntomas-diagnóstico (meses) | 14 [5-31] |
| Seguimiento (meses) | 34,5 [12-58] |
| Motivo de consulta | |
| Clínica extracardiaca | 69 (38,1) |
| Disnea | 39 (21,5) |
| Estudio familiar | 23 (12,7) |
| Síncope | 11 (6,1) |
| Mutaciones | |
| Val50Met | 120 (67,8) |
| Val142Ile | 22 (12,4) |
| Glu109Lys | 14 (7,8) |
| Ser97Tyr | 6 (3,4) |
| Val122Del | 4 (2,2) |
| Ser43Asn | 2 (1,1) |
| Asp38Asn | 2 (1,1) |
| Val71Ala | 1 (0,5) |
| Glu89Gln | 1 (0,5) |
| Ala65Thr | 1 (0,5) |
| Thr80Ala | 1 (0,5) |
| Leu32Pro | 1 (0,5) |
| Glu74Gln | 1 (0,5) |
| Ala128Val | 1 (0,5) |
Los valores expresan n (%) o mediana [intervalo intercuartílico].
Las mutaciones de tipo missense fueron las más frecuentes (97,7%). Se comunicaron 15 mutaciones diferentes, las más frecuentes fueron Val50Met (n=120; 67,8%) y Val142Ile (n=22; 12,4%). De estos últimos hay que destacar que solo 1 paciente era de raza negra (tabla 3). Si se descartan los 2 focos endémicos de Val50Met, en el resto de España la mutación Val50Met sigue siendo la más frecuente (n=60; 51,2%) (tabla 2).
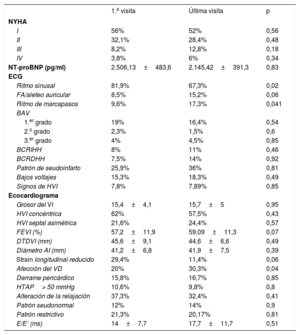
Características clínicas y exploratorias basales y al seguimiento
| 1.a visita | Última visita | p | |
|---|---|---|---|
| NYHA | |||
| I | 56% | 52% | 0,56 |
| II | 32,1% | 28,4% | 0,48 |
| III | 8,2% | 12,8% | 0,18 |
| IV | 3,8% | 6% | 0,34 |
| NT-proBNP (pg/ml) | 2.506,13±483,6 | 2.145,42±391,3 | 0,83 |
| ECG | |||
| Ritmo sinusal | 81,9% | 67,3% | 0,02 |
| FA/aleteo auricular | 8,5% | 15,2% | 0,06 |
| Ritmo de marcapasos | 9,6% | 17,3% | 0,041 |
| BAV | |||
| 1.er grado | 19% | 16,4% | 0,54 |
| 2.o grado | 2,3% | 1,5% | 0,6 |
| 3.er grado | 4% | 4,5% | 0,85 |
| BCRIHH | 8% | 11% | 0,46 |
| BCRDHH | 7,5% | 14% | 0,92 |
| Patrón de seudoinfarto | 25,9% | 36% | 0,81 |
| Bajos voltajes | 15,3% | 18,3% | 0,49 |
| Signos de HVI | 7,8% | 7,89% | 0,85 |
| Ecocardiograma | |||
| Grosor del VI | 15,4±4,1 | 15,7±5 | 0,95 |
| HVI concéntrica | 62% | 57,5% | 0,43 |
| HVI septal asimétrica | 21,6% | 24,4% | 0,57 |
| FEVI (%) | 57,2±11,9 | 59,09±11,3 | 0,07 |
| DTDVI (mm) | 45,6±9,1 | 44,6±6,6 | 0,49 |
| Diámetro AI (mm) | 41,2±6,8 | 41,9±7,5 | 0,39 |
| Strain longitudinal reducido | 29,4% | 11,4% | 0,06 |
| Afección del VD | 20% | 30,3% | 0,04 |
| Derrame pericárdico | 15,8% | 16,7% | 0,85 |
| HTAP> 50 mmHg | 10,6% | 9,8% | 0,8 |
| Alteración de la relajación | 37,3% | 32,4% | 0,41 |
| Patrón seudonormal | 12% | 14% | 0,9 |
| Patrón restrictivo | 21,3% | 20,17% | 0,81 |
| E/E’ (ms) | 14±7,7 | 17,7±11,7 | 0,51 |
AI: aurícula izquierda; BAV: bloqueo auriculoventricular; BCRDHH: bloqueo completo de la rama derecha del haz de His; BCRIHH: bloqueo completo de la rama izquierda del haz de His; DTDVI: diámetro telediastólico del ventrículo izquierdo; ECG: electrocardiograma; FA: fibrilación auricular; FEVI: fracción de eyección del ventrículo izquierdo; HTAP: hipertensión arterial pulmonar; HVI: hipertrofia ventricular izquierda; NT-proBNP: fracción aminoterminal del propéptido natriurético cerebral; NYHA: clase funcional de la New York Heart Association; VD: ventrículo derecho; VI: ventrículo izquierdo.
En la primera visita, la mayoría de los pacientes estaban en clase funcional de la New York Heart Association I o II y hasta un 12%, en clase funcional III o IV. La media de la fracción aminoterminal del propéptido natriurético cerebral (NT-proBNP) fue de 2.506±483 pg/ml. No hubo diferencias significativas en la clase funcional ni en los valores de NT-proBNP en el seguimiento. Respecto al electrocardiograma, en la primera visita el 25,3% presentaba algún tipo de bloqueo auriculoventricular, el 8,5% estaba en fibrilación auricular o aleteo y el 9,6% presentaba ritmo de marcapasos. El patrón de seudoinfarto estaba presente en el 25,9% de los electrocardiogramas y en el 15,3% había bajos voltajes. El 7,8% de los pacientes tenían signos de hipertrofia ventricular izquierda. En la última visita había aumentado el porcentaje de los que estaban en ritmo de marcapasos (el 17,3 frente al 9,6%; p=0,041) y el patrón de seudoinfarto (el 25,9 frente al 36%; p=0,81). En el ecocardiograma, el grosor ventricular medio fue de 15,4±4,1mm, y en el 62% la hipertrofia ventricular era concéntrica. La fracción de eyección del ventrículo izquierdo fue del 57,2%±11,9%. La media del diámetro anteroposterior de la aurícula izquierda fue 41,2±6,8mm. El strain longitudinal estaba reducido en segmentos basales respecto a los apicales en el 29,4% de los casos. El 15,8% de los pacientes tenían derrame pericárdico, leve en la mayoría (87,9%). Un 21,3% presentaba patrón diastólico restrictivo. En la última visita se observó mayor afección del ventrículo derecho (el 20 frente al 30,3%; p=0,04) (tabla 3).
De los pacientes sometidos a cardiorresonancia (n=71), se observó un patrón subendocárdico difuso en el 52,7% y focal en el 16,3%. La técnica de mapeo en T1 se empleó poco, solo en 10 pacientes, y en la mitad de ellos se observaba T1 nativo prolongado.
En la gammagrafía cardiaca con bisfosfonatos (n=80), se observó captación de grado 2-3 según la escala de Perugini en el 75% de los casos. Un 18,75% presentaban captación de grado 1 y el 6,25%, de grado 0.
Se realizó biopsia en alguna localización a 93 pacientes (52,5%). De las biopsias positivas, 23 (27,4%) fueron de grasa subcutánea; 19 (22,6%), endomiocárdicas; 15 (17,9%), de mucosa rectal y 15 (17,9%), del nervio sural.
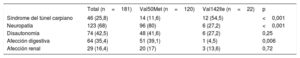
La afección extracardiaca fue frecuente: neurológica en el 68%, digestiva en el 35,4% y renal en el 16,4%. El 25,8% tenía antecedente de síndrome del túnel carpiano y el 42,5%, signos de disautonomía. La neuropatía y la afección digestiva fueron más frecuentes en los pacientes con mutación Val50Met (el 80 frente al 27,2%; p <0,001) que con Val142Ile (el 39,1 frente al 4,5%, p=0,006), mientras que el síndrome del túnel carpiano fue más frecuente en los portadores de la mutación Val142Ile que en la Val50Met (el 54,5 frente al 11,6%; p <0,001) (tabla 4).
Afección extracardiaca en la primera evaluación
| Total (n=181) | Val50Met (n=120) | Val142Ile (n=22) | p | |
|---|---|---|---|---|
| Síndrome del túnel carpiano | 46 (25,8) | 14 (11,6) | 12 (54,5) | <0,001 |
| Neuropatía | 123 (68) | 96 (80) | 6 (27,2) | <0,001 |
| Disautonomía | 74 (42,5) | 48 (41,6) | 6 (27,2) | 0,25 |
| Afección digestiva | 64 (35,4) | 51 (39,1) | 1 (4,5) | 0,006 |
| Afección renal | 29 (16,4) | 20 (17) | 3 (13,6) | 0,72 |
Los valores expresan n (%).
Se implantó un marcapasos a un tercio de los pacientes (34,3%). En la mitad de los casos (49,2%) la indicación fue por bloqueo auriculoventricular completo; en el 23,1%, por profilaxis antes del trasplante hepático y en el 18,5%, por disfunción sinusal. La prescripción de desfibrilador implantable no fue frecuente y solo se implantaron al 4% de los pacientes.
Respecto al tratamiento médico basal, el 41,5% estaban con diuréticos; el 30,2%, con inhibidores de la enzima de conversión de la angiotensina; el 17,5%, con bloqueadores beta; el 11,9%, con antagonistas de la aldosterona y el 20,9%, con anticoagulantes. Respecto a los tratamientos específicos para la ATTRv, el 25,4% estaba en tratamiento con tafamidis y el 24%, con diflunisal; el 31,6% había recibido trasplante hepático y el 4,5%, un trasplante cardiaco.
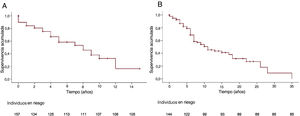
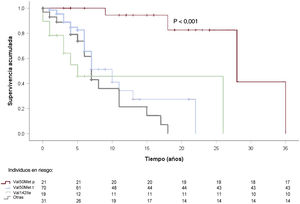
Durante una mediana de seguimiento de 34,5 meses, el 60,2% precisó ingreso hospitalario principalmente por causa cardiaca (59,6%) y neurológica (17,4%): en los primeros la mayoría fue por insuficiencia cardiaca (67%) y en los segundos, por ictus (57,8%). Fallecieron un tercio de los pacientes (32,5%), principalmente por insuficiencia cardiaca (28,8%), muerte súbita (11,5%) o causa neurológica o renal (17,3%). En la figura 1 y la figura 2 se muestran los análisis de la supervivencia.
 y desde el inicio de los síntomas hasta un final combinado de muerte o trasplante cardiaco (B). Medianas de 8 y 11 años respectivamente.")
, Val50Met (inicio tardío), Val142Ile y el resto, desde el inicio de los síntomas hasta un final combinado de muerte o trasplante cardiaco. Medianas de 28, 10, 5 y 7 años respectivamente.")
Curvas de supervivencia de los pacientes con amiloidosis hereditaria por transtirretina según genotipo: Val50Met (inicio precoz), Val50Met (inicio tardío), Val142Ile y el resto, desde el inicio de los síntomas hasta un final combinado de muerte o trasplante cardiaco. Medianas de 28, 10, 5 y 7 años respectivamente.
Se presenta el primer estudio que describe en una muestra amplia y representativa las principales características de los pacientes con ATTRv en España. Hasta la fecha se han publicado pocos datos, y de manera aislada, de solo 2 centros españoles5,9.
El diagnóstico de la afección cardiaca en la ATTRv ha mejorado en los últimos años merced a la posibilidad de un diagnóstico no invasivo mediante gammagrafía ósea y cardiorresonancia12. Por otra parte, la aparición de tratamientos específicos ha aumentado el interés el diagnóstico precoz.
En general, la mutación más frecuente en nuestro país es con diferencia la Val50Met (67,8%), no solo en los 2 focos endémicos de esta mutación (Mallorca y Huelva), sino también en el resto de España, aunque menos representada (51,2%). Existe un amplio abanico de mutaciones en España, un total de 15 variantes (tabla 2), de las que Val142Ile (la más frecuente en buena parte de Europa y Estados Unidos) es la segunda más frecuente (18,8%).
Históricamente la mutación Val50Met se consideraba casi puramente neurológica4. Nuestro estudio pone de manifiesto que la afección cardiaca en estos pacientes es frecuente. En el registro THAOS13, se describe que el fenotipo cardiaco en Europa se debe principalmente a las consideradas mutaciones cardiacas (Val142Ile, Leu131Met y Ile88Leu) y en Estados Unidos, a Val142Ile14, pero hay que tener en cuenta que la afección cardiaca con Val50Met también es considerable.
La presentación de la ATTRv es muy heterogénea, la mayoría de los pacientes presentan hipertrofia ventricular izquierda concéntrica y crecimiento auricular. Este estudio pone de manifiesto algunas de las claves para sospechar esta afección en los pacientes: insuficiencia cardiaca con fracción de eyección conservada, patrón de seudoinfarto, bajos voltajes o trastornos de la conducción, afección del ventrículo derecho, derrame pericárdico ligero o un gradiente de strain longitudinal entre la base y el ápex, entre otros. Desde el punto de vista clínico, se debe prestar atención a la afección extracardiaca que estará presente en la mayoría de los pacientes, sobre todo la neuropatía y la afección digestiva en los pacientes con Val50Met y el síndrome del túnel carpiano en aquellos con Val142Ile. Este fenotipo mixto ya se mostró como frecuente en la cohorte europea del registro THAOS13, si bien en nuestro estudio se demuestra que la afección extracardiaca es todavía mayor que lo publicado, por lo que cabe subrayar la importancia de hacer una valoración multidisciplinaria de los pacientes con ATTRv que integre diferentes especialidades.
A pesar del aumento en el número de diagnósticos de ATTRv con afección cardiaca, aún hay posibilidad de mejora, ya que el tiempo entre el inicio de los síntomas y el diagnóstico es de 1,9±2,3 años. Retrasos similares se describen en otros estudios, hasta 5 años de retraso en los pacientes sin historia familiar15.
En nuestro estudio se encontraron 5 pacientes (un 6% de los pacientes a los que se había hecho gammagrafía) con captación de grado 0 de la escala de Perugini. El protocolo actual para el diagnóstico de AC indica que es necesaria una gammagrafía con captación de grado 2-3; sin embargo, se han publicado estudios en los que, sobre todo en pacientes con Val50Met e inicio precoz y síntomas predominantemente neurológicos, esta prueba tiene baja sensibilidad (hasta un 41%)11,16. En nuestra serie todos estos pacientes presentaban afección neurológica, 3 de ellos tenían la mutación Val50Met y 2, la Ser97Tyr con afección neurológica. Estos 2 últimos tenían una cardiorresonancia con realce tardío de gadolinio subendocárdico difuso compatible con el diagnóstico de AC y, de los 3 casos con la mutación Val50Met que no tenían ningún grado de captación, en 2 había una hipertrofia ventricular no explicable por otras causas junto con trastornos de la conducción (bloqueo auriculoventricular de primero y segundo grado Mobitz I respectivamente) y en el otro, bloqueo auriculoventricular completo sincopal y marcapasos con menos de 60 años.
Respecto al tratamiento, hay que los bloqueadores beta, los inhibidores de la enzima de conversión de la angiotensina y otros tratamientos estándar de la insuficiencia cardiaca son frecuentes en nuestra serie, pero es conocido que se deben administrar con precaución, ya que suelen tolerarse mal y pueden ser perjudiciales, especialmente los bloqueadores beta17. Asimismo, solo 1 de cada 5 pacientes de nuestra serie recibía anticoagulación. La AC presenta alta prevalencia de trombos intracardiacos y embolias que contribuyen a aumentar la mortalidad, por lo que la estratificación del riesgo embolígeno debe hacerse con cuidado y, en general, se recomienda anticoagular precozmente18. Se piensa que en algunos centros se utilizaron erróneamente los criterios basados en CHA2DS2-VASc. En nuestra serie la prevalencia de fibrilación auricular/aleteo auricular fue solo del 8,5%, que aumentaba al 15,2% en el seguimiento, y el tamaño basal de la aurícula izquierda era de 41,2±6,8 mm y tras el seguimiento, 41,9±7,5mm. Se piensa que estos porcentajes, junto con lo ya comentado, influyó en la decisión de los médicos responsables de no anticoagular a más pacientes.
En nuestra serie se observó una alta mortalidad, y las causas cardiacas (insuficiencia cardiaca y muerte súbita) fueron las principales. A pesar de ello, el implante de desfibriladores no es frecuente, probablemente porque en muchos casos la afección extracardiaca condiciona negativamente el pronóstico de los pacientes o porque la cardiopatía se encuentra en una fase muy avanzada. Por otra parte, no está claramente demostrado el beneficio del desfibrilador en prevención primaria19. Las elevadas tasas de descompensación por insuficiencia cardiaca y de trasplante cardiaco y la elevada mortalidad nos hacen pensar que el diagnóstico se hace en una fase avanzada de la enfermedad y que se debería mejorar el diagnóstico precoz de esta afección para poder aplicar los tratamientos que han demostrado beneficio en etapas tempranas.
Destaca del análisis de supervivencia que las mutaciones diferentes de Val50Met se asocian en general con peor pronóstico y que, a su vez, la mutación Val50Met de inicio precoz (< 50 años) (fenotipo más neurológico) tiene un mejor pronóstico que las demás mutaciones (Val50Met de inicio tardío, Val142Ile y otras) (fenotipo más cardiológico), lo que va en la línea de otros artículos, que muestran que las mutaciones con fenotipo predominante cardiaco tienen un peor pronóstico20. Asimismo, hay que destacar de los análisis de supervivencia la potencial mejora del pronóstico de estos pacientes si no transcurriesen tantos años desde el inicio de los síntomas hasta el diagnóstico.
Por último, más del 80% de los pacientes habían recibido algún tratamiento específico de la ATTRv, ya sea trasplante hepático, tafamidis o diflunisal, lo que habrá supuesto en muchos casos un impacto beneficioso significativo en su evolución. Respecto a la cuarta parte de la serie total que había recibido tratamiento con tafamidis, este se prescribió por indicación neurológica (dosis de 20mg/día) y no para tratamiento de la cardiopatía, ya que los datos recogidos en este estudio son anteriores a la publicación del estudio ATTR-ACT6 en el que se utilizó una dosis muy superior. Asimismo, hay que destacar que los datos de este estudio se recogieron en 2018-2019, y en ese momento la Agencia Española del Medicamento aún no había aprobado los fármacos inhibidores de la síntesis de TTR (patisirán e inotersén).
CONCLUSIONESLa ATTRv presenta frecuentemente afección cardiaca. En este estudio se describen las principales características de esta enfermedad en la población española. Existe un amplio abanico de mutaciones, si bien la más frecuente es Val50Met. La afección extracardiaca es frecuente, sobre todo la neuropatía y el síndrome del túnel carpiano. En el momento actual, en que es posible evaluar la afección cardiaca de manera no invasiva, se debe conocer esta enfermedad de presentación heterogénea para diagnosticarla y tratarla precozmente.
FINANCIACIÓNEl presente trabajo ha recibido financiación mediante la concesión de una beca de investigación de la Sección de Insuficiencia Cardiaca de la Sociedad Española de Cardiología del año 2018.
CONTRIBUCIÓN DE LOS AUTORESDiseño del estudio y creación de la base de datos: Tomás Ripoll-Vera, Jorge Álvarez Rubio. Selección de pacientes e inclusión de variables en la base de datos: Jorge Álvarez Rubio, Ana José Manovel Sánchez, José González-Costello, Pablo García-Pavía, Javier Limeres Freire, José Manuel García-Pinilla, Esther Zorio Grima, Ana García-Álvarez, María Valverde Gómez, M. Ángeles Espinosa Castro, Gonzalo Barge-Caballero, Juan Ramón Gimeno Blanes, María Teresa Bosch Rovira, Luis Miguel Rincón Díaz, Miguel Ángel Aibar Arregui, María Gallego-Delgado, Juan Jiménez-Jáimez, Marina Martínez Moreno, Mayte Basurte, Xabier Arana Achaga, Idaira Famara Hernández Baldomero. Análisis de resultados: Jorge Álvarez Rubio, Tomás Ripoll-Vera. Redacción del manuscrito: Jorge Álvarez Rubio, Tomás Ripoll-Vera. Revisión crítica del manuscrito: José González-Costello, Ana José Manovel Sánchez, Pablo García-Pavía, Javier Limeres Freire, José Manuel García-Pinilla, Esther Zorio Grima, Ana García-Álvarez, María Valverde Gómez, M. Ángeles Espinosa Castro, Gonzalo Barge-Caballero, Juan Ramón Gimeno Blanes, María Teresa Bosch Rovira, Luis Miguel Rincón Díaz, Miguel Ángel Aibar Arregui, María Gallego-Delgado, Juan Jiménez-Jáimez, Marina Martínez Moreno, Mayte Basurte, Xabier Arana Achaga, Idaira Famara Hernández Baldomero.
CONFLICTO DE INTERESESJ. González-Costello ha recibido honorarios por ponencias en congresos y cursos de Pfizer y Alnylam, ha participado en consultorías de Pfizer y Alnylam y es presidente actual de la Asociación de Insuficiencia Cardiaca de la Sociedad Española de Cardiología. E. Zorio Grima ha recibido honorarios por ponencias en congresos y cursos de Alnylam. A. García-Álvarez ha recibido honorarios por ponencias en congresos y cursos de Novartis, Rovi y AstraZeneca y tiene una patente actualmente. G. Barge-Caballero ha recibido pago por una beca de investigación, ha recibido honorarios por ponencias en congresos y cursos de Pfizer, AstraZeneca, Novartis y Servier y ha participado en consultorías de Pfizer. M. Basurte ha recibido honorarios por consultoría de Pfizer. T. Ripoll-Vera obtuvo pago por beca de investigación de la Asociación de Insuficiencia cardiaca de la Sociedad Española de Cardiología. El resto de los autores no declaran conflictos de intereses.
AGRADECIMIENTOSA Catalina Meliá Mesquida y Yolanda Gómez Pérez por su labor en la coordinación de pacientes y la gestión de la investigación en la Unidad de Cardiopatías Familiares, y al Grupo Multidisciplinar de ATTR del Hospital Universitario de Son Llàtzer.
Ainara Lozano Bahamonde (Hospital Universitario Basurto), Coloma Tirón (Hospital Universitari Dr. Josep Trueta, Girona), José Onaindia Gandarias y Ángela Cacicedo Fernández de Bobadilla (H. Galdakao, Bizkaia), Pablo Elpidio García Granja y Javier López Díaz (Hospital Clínico Universitario Valladolid), Amaria Núñez Íñiguez (Hospital Universitario Cruces, Bilbao), Mercedes Rivas-Lasarte (Hospital Santa Creu i Sant Pau, Barcelona), Sonia Ruiz Bustillo (Hospital del Mar, Barcelona), Juan Caro (Hospital Universitario La Paz, Madrid), Marta Padilla-Sainz, Carlos Casasnovas, Carmen Baliellas, Laura Lladó, Emma González-Vilatarsana y Carles Díez-López (Hospital Universitari de Bellvitge, Barcelona), José F. Rodríguez-Palomares (Hospital Universitario Vall d’Hebron, Barcelona), Ainhoa Robles-Mezcua y Arancha Díaz-Expósito (Hospital Universitario Virgen de la Victoria, Málaga), Javier Navarrete-Navarro (Hospital Universitario y Politécnico La Fe, Valencia), José T. Ortiz-Pérez (Hospital Clínic-IDIBAPS, Barcelona), Julián Palomino-Doza y Rafael Salguero-Bodes (Hospital Universitario 12 de Octubre, Madrid; CIBERCV), Irene Méndez Fernández (Hospital Gregorio Marañón, Madrid), Roberto Barriales-Villa y José M. Larrañaga-Moreira (Complexo Hospitalario Universitario de A Coruña), Elena Fortuny Frau y Jaume Pons Llinares (Hospital Universitario Son Espases, Palma de Mallorca), Pablo Revilla Martí (Hospital Clínico Universitario de Zaragoza, IIS-A, Zaragoza), Eduardo Villacorta Argüelles (Complejo Asistencial Universitario de Salamanca, Salamanca), Rosa Macías Ruiz (Hospital Universitario Virgen de las Nieves, Granada), Irene Rilo Miranda e Itziar Solla Ruiz (Hospital Universitario Donostia, San Sebastián), Antonio Lara Padrón y Francisco Bosa Ojeda (Complejo Hospitalario Universitario de Canarias), Paula Morlanes Gracia y Guido Antoniutti (Hospital Universitario Son Llàtzer, Palma de Mallorca).
- –
La ATTRv es una enfermedad multisistémica con una prevalencia mayor de lo que se pensaba antiguamente, y está claramente infradiagnosticada.
- –
El diagnóstico es tardío debido al escaso conocimiento por la comunidad médica y la variable expresión de la enfermedad.
- –
En los últimos años se ha avanzado considerablemente en el diagnóstico no invasivo y en nuevas moléculas que mejoran el pronóstico de la enfermedad.
- –
El perfil genotípico y fenotípico de la ATTRv general en España hasta ahora era desconocido.
- –
La presentación clínica y las características de los pacientes con ATTRv en España es heterogénea y difiere sustancialmente según el genotipo; la afección cardiaca es muy frecuente.
- –
En España, el diagnóstico de ATTRv se realiza de manera no invasiva cada vez con mayor frecuencia.