

Las canalopatías cardiacas son trastornos genéticos que pueden causar muerte súbita. Entre ellas se encuentran el síndrome de QT largo y el síndrome de Brugada. Ambos se diagnostican según unos criterios previamente publicados. Nuestro objetivo es evaluar la sensibilidad de esos criterios en una serie consecutiva de sujetos portadores de mutación patogénica para síndrome de QT largo y síndrome de Brugada.
MétodosSe incluyó a 15 familias y 31 sujetos portadores de mutaciones con alta probabilidad patogénica de síndrome de QT largo o síndrome de Brugada. Realizamos estudio clínico y electrocardiográfico para analizar el cumplimiento de los criterios diagnósticos. El estudio estadístico se realizó con el programa estadístico SPSS 17.0.
ResultadosEl 48,3% de los sujetos cumplían criterios de alta probabilidad de síndrome de QT largo o síndrome de Brugada. Entre la población con mutación para síndrome de QT largo, sólo 10 de 21 sujetos mostraron un índice de Schwartz ≥ 4. Tanto la mediana de la puntuación de Schwartz como el intervalo QTc fueron menores en familiares que en probandos. En la población con mutación para síndrome de Brugada, el 60% no cumplía los criterios diagnósticos vigentes, algo que fue más frecuente en familiares. El test farmacológico con epinefrina y flecainida ayudó a establecer el diagnóstico en dos familias portadoras de mutación con fenotipo negativo.
ConclusionesLos criterios diagnósticos actuales para síndrome de QT largo y síndrome de Brugada tuvieron baja sensibilidad en nuestra muestra de portadores genéticos. El test genético apoyado por tests farmacológicos puede incrementar la sensibilidad diagnóstica, especialmente en familiares asintomáticos.
Palabras clave
La muerte súbita en sujetos con corazón estructuralmente normal (síndrome de la muerte súbita arrítmica) suele ser de origen arrítmico y está causada predominantemente por canalopatías o trastornos de los canales iónicos cardiacos1. Entre estas se incluyen como más prevalentes el síndrome de QT largo (SQTL), el síndrome de Brugada (SB), la taquicardia ventricular catecolaminérgica polimórfica y la fibrilación ventricular idiopática (FVI)2. Las características comunes de las canalopatías son el origen genético definido, corazón estructuralmente normal, predisposición a las arritmias ventriculares y la muerte súbita y una penetrancia clínica habitualmente incompleta3. La evaluación cardiológica exhaustiva con electrocardiograma (ECG) de superficie y tests farmacológicos con flecainida y epinefrina consigue identificar la etiología subyacente a un episodio arrítmico en una proporción de casos sustancial4–6. Los casos de muerte súbita en que no se llega a un diagnóstico final pese a realizarse las pruebas convencionales se catalogan de FVI.
Para el diagnóstico de las canalopatías, y más concretamente del SQTL y el SB, existen unos criterios diagnósticos propuestos y vigentes hasta la fecha (tabla 1). Los criterios de Schwartz, publicados en 1985 y modificados en 19937, consideran el diagnóstico de SQTL como probable cuando la puntuación alcanza 4 o más puntos y tienen en cuenta datos electrocardiográficos, clínicos y de historia familiar (tabla 1A). Para el SB, los criterios vigentes desde 20058 exigen la presencia del patrón de Brugada tipo 1 en al menos dos derivaciones precordiales del ECG de superficie basal o tras la infusión intravenosa de un bloqueador del sodio (tabla 1B).
Criterios diagnósticos del síndrome de QT largo y de Brugada
| Puntos | |
| A) Criterios diagnósticos de SQTL | |
| Hallazgos en ECG | |
| QTc calculado mediante la fórmula de Bazzet>480 ms | 3 |
| 460-40 ms | 2 |
| 450 ms (varones) | 1 |
| Torsade de pointes | 2 |
| Alternancia de la onda T | 1 |
| Onda T bífida en tres derivaciones | 1 |
| Frecuencia cardiaca basal<2 centiles de lo correspondiente para la edad | 0,5 |
| Historia clínica | |
| Síncope con estrés | 2 |
| Síncope sin estrés | 1 |
| Sordera congénita | 0,5 |
| Historia familiar | |
| Familiares de primer grado con SQTL definitivo por criterios de Schwartz | 1 |
| Familiares de primer grado con muerte súbita antes de los 30 años | 0,5 |
| B) Criterios diagnósticos de SB | |
| Criterio en ECG (necesario) | Uno de los siguientes debe estar presente |
| Patrón de Brugada tipo 1 en dos o más derivaciones (V1-V3) en presencia o ausencia de un bloqueador de los canales de Na | FV documentada |
| TV polimórfica documentada | |
| Historia familiar de muerte súbita en menores de 40 años | |
| Patrón ECG tipo 1 en familiares directos | |
| TV inducible en EEF | |
| Síncope | |
| Respiración agónica nocturna | |
ECG: electrocardiograma; EEF: estudio electrofisiológico; FV: fibrilación ventricular; SB: síndrome de Brugada; SQTL: síndrome de QT largo; TV: taquicardia ventricular.
SQTL≥4 puntos: probabilidad alta; SQTL de 2-3 puntos: probabilidad intermedia; SQTL≤1 punto: probabilidad baja.
Ambos criterios diagnósticos no tienen en cuenta la información del estudio genético. Estudios recientes indican la importancia del estudio genético combinado con test farmacológico dirigido por fenotipo para llegar al diagnóstico en casos dudosos debido a la penetrancia clínica incompleta de estos trastornos, sobre todo en familiares asintomáticos9–13. El objetivo de nuestro trabajo es analizar la validez de los criterios diagnósticos en sujetos con diagnóstico de canalopatía altamente probable con base en el resultado del test genético, y estudiar su valor diagnóstico en probando y familiares, apoyado por el test farmacológico en pacientes portadores de mutación genética y fenotipo negativo.
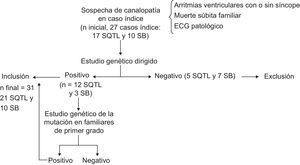
MÉTODOSSe estudió a los casos índice sospechosos de padecer una canalopatía por presentar una o varias de las siguientes características: haber sufrido arritmias ventriculares polimórficas de origen no filiado o síncope de perfil cardiogénico, presentar un ECG que apunte a SQTL o SB o ser familiar directo de una persona fallecida de muerte súbita probablemente arrítmica o por síndrome de muerte súbita arrítmica (autopsia sin hallazgos estructurales y contexto de la muerte que indique muerte súbita cardiaca). Se sometió a todos ellos a estudio genético para secuenciar los genes más prevalentes en SQTL y SB. Se incluyó en el estudio sólo a los probandos portadores de una mutación patogénica. A los familiares directos de estos pacientes con mutación, se los estudió con ECG y test genético para buscar la misma mutación encontrada en el probando. Se incluyó en el estudio a los familiares portadores de mutación, y se excluyó a los no portadores y los casos con causa secundaria y reversible de prolongación del intervalo QTc (fig. 1).

Algoritmo de inclusión de pacientes. Se excluyó a los casos índice y los familiares con genotipo negativo. Nótese que la prevalencia mutacional fue similar a las de estudios previos, con un 70,5% para síndrome de QT largo y un 30% para síndrome de Brugada. ECG: electrocardiograma; SB: síndrome de Brugada; SQTL: síndrome de QT largo.
La mutación hallada debía estar asociada con alta probabilidad a SQTL o SB, bien por haber sido descrita previamente, bien por presentar características moleculares que hagan que se la considere de alta probabilidad patogénica, como la localización en la proteína, el cambio aminoacídico resultante o efectos descritos de mutaciones en zonas cercanas del mismo gen14.
Se sometió a todos los sujetos a estudio con ECG a 25 y 50mm/s, midiendo el intervalo QTc por la fórmula de Bazzet15 y buscando signos de patrón de Brugada tipo 1 (elevación de segmento ST en al menos dos derivaciones precordiales de más de 1 mm con morfología típica). A todos se les practicó ecocardiografía transtorácica para descartar cardiopatía estructural. En los casos sin fenotipo en el ECG, se realizaron tests farmacológicos con epinefrina y/o flecainida13,16 orientados en función de genotipo y/o factores epidemiológicos de sospecha (muerte súbita en familiar en reposo para el SB o situación de máximo esfuerzo para el SQTL, estrés físico o emocional desencadenante de síncopes para el SQTL, síncopes o arritmias ventriculares en respuesta a estímulos auditivos o en relación con el baño para diferentes subtipos de SQTL, etc.). Se calculó el índice de Schwartz de los sujetos portadores de mutación en genes causantes de SQTL. Se evaluó de manera dicotómica si se cumplían criterios diagnósticos de SB.
Estudio estadísticoEl estudio estadístico se realizó con el programa SPSS 17.0 (Chicago, Estados Unidos). Se estudió con variables descriptivas la proporción de sujetos que cumplían criterios diagnósticos de cada enfermedad (medidas de tendencia central, como la media y la mediana, y de proporción, como el porcentaje). Posteriormente se realizó comparación estadística mediante tests no paramétricos (U de Mann-Whitney y test exacto de Fisher) entre probando y familiares directos para buscar diferencias en cuanto a variables de expresión fenotípica.
El estudio ha sido aprobado por el comité de ética de nuestro hospital y todos los pacientes firmaron el consentimiento informado para el estudio con secuenciación genética.
RESULTADOSSe evaluaron inicialmente 27 casos índice con sospecha de canalopatía (fig. 1); en 17 casos se sospechaba SQTL y en los otros 10, SB. Se excluyó del estudio a 5 casos de SQTL por no haber confirmación genética del diagnóstico (prevalencia mutacional del 70,5%), y 7 casos índice con diagnóstico clínico de SB no mostraron mutaciones en SCN5A, por lo que también se los excluyó (prevalencia mutacional del 30%). De los 12 casos índice con SQTL, el motivo de consulta había sido síncope en 6 y parada cardiaca por fibrilación ventricular en 4; 2 casos se diagnosticaron tras estudio por muerte súbita cardiaca en familiar directo. De los 3 probandos con SB incluidos, 2 habían sufrido parada cardiaca y 1 estaba asintomático.
Las mutaciones halladas en los pacientes con SQTL se localizaban principalmente en el gen KCNH2; encontramos una mutación en KCNQ1 y en 2 casos el gen afectado era el SCN5A. Estas mutaciones habían sido descritas previamente por nuestro grupo en un trabajo reciente10,17. En el caso del SB, las tres mutaciones halladas no estaban previamente descritas y se describen en la tabla 2. De todos los casos positivos, en 2 se había realizado el estudio genético por FVI con ECG y tests farmacológicos normales.
El 20% de las mutaciones halladas estaban previamente descritas como asociadas a SQTL, mientras que en otros 2 casos nuestro grupo realizó un estudio de las propiedades electrofisiológicas del canal mutado en un modelo celular. En el resto de las mutaciones, nos apoyamos en los datos anteriormente expuestos para establecer su patogenicidad14,17.
Incluyendo a los familiares de primero y segundo grado que mostraron la misma mutación altamente sospechosa de patogenicidad, se alcanzó finalmente un tamaño muestral de 31 sujetos portadores de mutaciones (21 SQTL, de los que 12 eran probandos y 9, familiares; 10 SB, 3 probandos y 7 familiares).
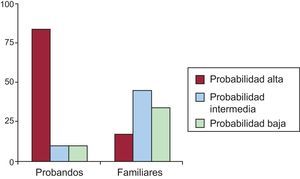
De acuerdo con los criterios mencionados anteriormente, 15 de los 31 sujetos analizados cumplían los criterios diagnósticos de la canalopatía en cuestión descritos en la tabla 1, lo que supone un 48,3% del total, 11 de 15 probandos (73,3%) y 4 de 16 familiares (25%). Por lo tanto, el estudio genético identificó la canalopatía subyacente en 4 probandos con fenotipo indeterminado y detectó el estado de portador silente en 12 familiares sin expresión fenotípica. A continuación y en las tablas 3 y 4, se exponen los resultados en función del tipo de canalopatía y el análisis comparativo de la expresión fenotípica entre probandos y familiares.
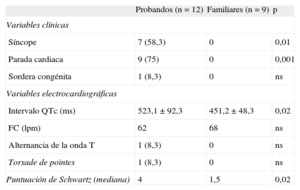
Evaluación fenotípica en portadores de mutación para síndrome de QT largo
| Probandos (n=12) | Familiares (n=9) | p | |
| Variables clínicas | |||
| Síncope | 7 (58,3) | 0 | 0,01 |
| Parada cardiaca | 9 (75) | 0 | 0,001 |
| Sordera congénita | 1 (8,3) | 0 | ns |
| Variables electrocardiográficas | |||
| Intervalo QTc (ms) | 523,1±92,3 | 451,2±48,3 | 0,02 |
| FC (lpm) | 62 | 68 | ns |
| Alternancia de la onda T | 1 (8,3) | 0 | ns |
| Torsade de pointes | 1 (8,3) | 0 | ns |
| Puntuación de Schwartz (mediana) | 4 | 1,5 | 0,02 |
FC: frecuencia cardiaca; ns: no significativo.
Los datos expresan media±desviación estándar o n (%).
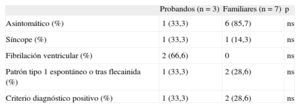
Evaluación fenotípica en portadores de mutación para síndrome de Brugada
| Probandos (n=3) | Familiares (n=7) | p | |
| Asintomático (%) | 1 (33,3) | 6 (85,7) | ns |
| Síncope (%) | 1 (33,3) | 1 (14,3) | ns |
| Fibrilación ventricular (%) | 2 (66,6) | 0 | ns |
| Patrón tipo 1 espontáneo o tras flecainida (%) | 1 (33,3) | 2 (28,6) | ns |
| Criterio diagnóstico positivo (%) | 1 (33,3) | 2 (28,6) | ns |
ns: no significativo.
De los 21 sujetos con genotipo de SQTL, tan sólo 12 (10 probandos y 2 familiares) cumplían criterios diagnósticos de Schwartz de alta probabilidad de padecer un SQTL (57,1%). Presentaban probabilidad intermedia para SQTL 2 (9,5%), y 7 (33,3%) mostraron baja probabilidad de padecer SQTL de acuerdo con lo criterios diagnósticos actuales. En la figura 2 se representa la diferencia entre probandos y familiares de la puntuación de Schwartz. La media de edad de este grupo era 24,1±17 años y el 41,7% eran varones. No hubo diferencias en la expresión fenotípica en función de edad o sexo.

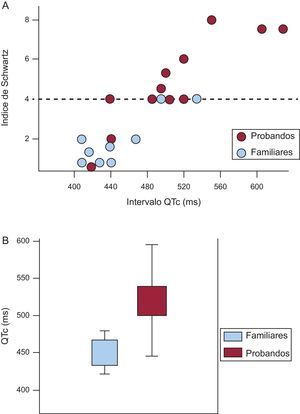
Como se aprecia en la figura 3A, esta alta proporción de sujetos que no cumplen criterios de SQTL muestra en general valores de intervalo QTc normales o en el límite superior de la normalidad, lo que hace que su puntuación final de Schwartz sea<4. El coeficiente de correlación rho de Spearman para ambas variables fue 0,89, es decir, alta correlación entre ambos parámetros. Analizando por subgrupos (fig. 3A), la proporción de casos que presenta puntuación de Schwartz<4 es significativamente mayor entre los familiares directos, habitualmente asintomáticos, que entre los casos índice. La mediana de la puntuación de Schwartz para el grupo de probandos fue 4, mientras que en el grupo de familiares fue 1,5 (p=0,02). La mayoría (7 de 9) de los familiares asintomáticos tenían un índice de Schwartz<4.
 y familiares (azul); se observa la buena correlación entre los valores del intervalo QTc y el índice de Schwartz; la línea de puntos representa el valor 4 del índice a partir del cual el diagnóstico de síndrome de QT largo es probable; nótese la gran cantidad de familiares que no alcanzan dicho valor y los 2 casos índice, ambos con historia de fibrilación ventricular, con puntuación<2. B: comparación de la longitud del intervalo QTc entre probando y familiares. Esta figura se muestra a todo color solo en la versión electrónica del artículo.")
A: gráfico representativo de la distribución de los intervalos QTc en probandos (rojo) y familiares (azul); se observa la buena correlación entre los valores del intervalo QTc y el índice de Schwartz; la línea de puntos representa el valor 4 del índice a partir del cual el diagnóstico de síndrome de QT largo es probable; nótese la gran cantidad de familiares que no alcanzan dicho valor y los 2 casos índice, ambos con historia de fibrilación ventricular, con puntuación<2. B: comparación de la longitud del intervalo QTc entre probando y familiares. Esta figura se muestra a todo color solo en la versión electrónica del artículo.
Los valores del intervalo QTc fueron los siguientes: el grupo de casos índice tuvo un intervalo QTc medio de 523,1±92,3 ms, mientras que en familiares era 451,2±48,3 ms (p<0,05) (fig. 3B). No todos los probandos mostraron intervalo QTc prolongado ni puntuación de Schwartz ≥ 4. De hecho, 2 casos índice que habían presentado episodio de fibrilación ventricular mostraron ECG basales normales y su puntuación de Schwartz indicaba baja probabilidad de SQTL. En estos 2 casos, el test de adrenalina fue negativo y fue el test genético lo que proporcionó el diagnóstico de SQTL.
Síndrome de BrugadaLa media de edad de este grupo era 43,5±12,5 años, y el 72,1% eran varones. No hubo diferencias en la expresión fenotípica en función de edad o sexo.
De los 10 casos portadores de mutación en SCN5A causante de SB, tan sólo 3 cumplían los criterios vigentes para el diagnóstico de esta canalopatía. Este porcentaje era mayor en los probandos que en los familiares directos (tabla 4). De los 3 casos índice, 2 no cumplían criterios diagnósticos al no mostrar el patrón de Brugada en ningún momento, ni siquiera tras flecainida intravenosa; uno de ellos se había presentado como fibrilación ventricular (familia 2 en la tabla 2). En este caso, el diagnóstico fue posible gracias al estudio genético y el test farmacológico en dos familiares directos que sí cumplían criterios diagnósticos, pues mostraron el patrón de Brugada tipo 1 tras flecainida (fig. 4). En el otro caso, el ECG presentaba el patrón tipo 1 en una sola derivación, por lo que tampoco cumplía los criterios diagnósticos.
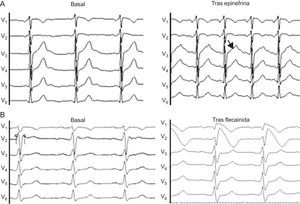
. A: corresponde a un familiar portador de mutación en KCNH2, hermano de un varón con muerte súbita y autopsia sin hallazgos patológicos; nótese la onda T mellada en su rama ascendente tras la infusión de adrenalina, signo característico de síndrome de QT largo tipo 216. B: test de flecainida en familiar directo de mujer afectada por fibrilación ventricular y electrocardiograma de superficie normal; el electrocardiograma de base ya indica síndrome de Brugada.")
Test farmacológico en casos dudosos (electrocardiograma a 50 mm/s). A: corresponde a un familiar portador de mutación en KCNH2, hermano de un varón con muerte súbita y autopsia sin hallazgos patológicos; nótese la onda T mellada en su rama ascendente tras la infusión de adrenalina, signo característico de síndrome de QT largo tipo 216. B: test de flecainida en familiar directo de mujer afectada por fibrilación ventricular y electrocardiograma de superficie normal; el electrocardiograma de base ya indica síndrome de Brugada.
Se estudió y se comparó los ECG basales de los 6 portadores genéticos en SCN5A que no mostraron el patrón de Brugada tipo 1 basal o tras flecainida con los de familiares directos de estas familias que no presentaban la mutación, y se halló que el intervalo PR (186,2±34 frente a 153,6±26 ms) y QRS (109,1±23 frente a 88,4±13 ms) estaban significativamente prolongados en los portadores de mutación en SCN5A respecto a los controles (p<0,05). No encontramos ningún ECG que mostrase el patrón de Brugada tipo 2, y el intervalo QTc fue similar entre ambos grupos.
Tests farmacológicosEn los probandos y/o familiares con ECG normal o que no cumplían criterios diagnósticos, se realizó test farmacológico con epinefrina y/o flecainida en función de la sospecha clínica. Se realizaron tres tests de epinefrina en portadores de mutación en genes de SQTL (2 en KCNH2 y 1 en SCN5A) y 8 tests de flecainida en portadores genéticos en SCN5A. De estos, el test de epinefrina ayudó a confirmar el diagnóstico en una familia con una mutación en KCNH2, todos asintomáticos y familiares de un sujeto con muerte súbita a los 25 años de edad (fig. 4A). En el caso del SB, el test de flecainida demostró el patrón tipo 1 en 2 familiares directos de la probando con FVI y una mutación en SCN5A, y el diagnóstico se confirmó en los 3 sujetos (fig. 4B). En el resto de los casos el test farmacológico resultó negativo.
DISCUSIÓNEl diagnóstico de los trastornos de los canales iónicos cardiacos en ocasiones no es sencillo. Para ello se establecieron hace años unos criterios diagnósticos que aún siguen vigentes (tabla 1)7,8. La observación de que numerosos casos de pacientes con diagnóstico genético confirmado de SQTL y SB no cumplían criterios diagnósticos no es reciente, si bien no hay ningún estudio en concreto que verifique su sensibilidad en una muestra genotipado de sujetos portadores de mutación patogénica3,18. En el caso del SB, ya se había señalado en un trabajo reciente que es necesaria una revisión para incluir los casos que muestren el patón tipo 1 en una sola derivación, con lo que aumentaría la sensibilidad diagnóstica en nuestra serie, al haber un probando genotipado que mostró el patrón en una sola derivación19.
Nosotros presentamos una serie de portadores de mutaciones patogénicas para SQTL y SB en los que hemos estudiado cada uno de los criterios diagnósticos. Nuestro principal hallazgo es que se identificaría a menos de la mitad de los portadores si no dispusiéramos del estudio genético apoyado en los tests farmacológicos. Este hecho ha sido más acentuado en familiares asintomáticos, pues en más del 70% de los casos no se cumplían criterios diagnósticos. Esto confirma que la penetrancia, es decir, la expresión del fenotipo en sujetos portadores de una mutación probablemente patogénica, es baja. Sin embargo, no a todos ellos se los puede considerar pacientes por el hecho de portar una mutación genética, ya que existen portadores que nunca expresan el fenotipo y otros sólo lo expresan en situaciones que alteren la repolarización, como toma de fármacos o trastornos iónicos (portadores silentes). Discernir qué portadores genéticos expresarán el fenotipo y cuáles no lo mostrarán es difícil, y en ese sentido los tests farmacológicos pueden ser un apoyo fundamental, tal y como muestra nuestro trabajo. Y es que la disfunción generada en el canal iónico puede ser de distinto grado en cada caso, y su repercusión funcional, además, depende de si puede ser compensada en parte o totalmente por otras corrientes iónicas. En este sentido, el hallazgo de que los familiares portadores genéticos de mutación en SCN5A, a pesar de tener fenotipo negativo para SB, muestran intervalos PR y QRS prolongados respecto a los familiares no portadores señala cierto grado de disfunción en el canal de sodio.
El hecho de padecer una canalopatía silente o con baja penetrancia no significa que no haya riesgo de arritmias ventriculares y muerte súbita20–22. En la serie de Priori et al3 se demostró que la penetrancia clínica en ciertas familias con muerte súbita familiar puede ser extremadamente baja para el SQTL. Asimismo, en el registro CASPER de FVI, el uso combinado de los tests farmacológicos y genéticos sirvió para desenmascarar una canalopatía subclínica en 24 de 63 pacientes con ECG basal normal9. En el trabajo que presentamos, 2 probandos con SQTL y 2 con SB no cumplían criterios diagnósticos, y 3 de ellos se habían presentado como FVI. Ello refuerza el valor diagnóstico del test genético en probandos sintomáticos en que los tests convencionales no consiguen identificar la canalopatía causante. Aunque parece que el intervalo QTc>500 ms y el patrón de Brugada espontáneo tipo 1 aumentan el riesgo de muerte súbita23,24, que no aparezcan estas alteraciones en el ECG de superficie reduce el riesgo arrítmico, pero no lo elimina por completo. Por ello, es relevante detectar estos casos de familiares asintomáticos para poder establecer medidas preventivas como la evitación de fármacos y situaciones potencialmente inductores de arritmias, y considerar el tratamiento con bloqueadores beta en el caso del SQTL.
La identificación de una mutación en un sujeto en quien sospechamos una canalopatía debe ser interpretada adecuadamente, sobre todo en el caso de familiares asintomáticos. Nuestro estudio indica que el test genético puede ser determinante para alcanzar el diagnóstico de una canalopatía oculta en probandos. Sin embargo, un hallazgo genético sin clara repercusión o cosegregación familiar con la enfermedad puede llevar a error14,25. Por ello, un equipo experto y conocedor de la genética cardiovascular debe evaluar e interpretar adecuadamente las mutaciones genéticas encontradas. Todas las mutaciones presentes en nuestra serie mostraban muy alta probabilidad de ser causantes de SQTL o SB. Esto lo razonamos por la posición del aminoácido afectado en la proteína, el tipo de cambio aminoacídico resultante y la alteración esperable de la estructura de la proteína14. En algunos casos, la mutación estaba descrita previamente en la lliteratura médica, a veces incluso con estudio in vitro de las propiedades electrofisiológicas del canal, y la conclusión sobre sus posibles efectos patogénicos en estos casos fue más evidente17,26,27.
Los criterios diagnósticos vigentes no tienen en cuenta los hallazgos genéticos a la hora de establecer la probabilidad de padecer la canalopatía en cuestión. En nuestra serie de portadores genéticos, la sensibilidad de los criterios vigentes actualmente no llegó al 50%, si bien, como ya se ha expuesto, probablemente no todos ellos sufrirán el fenotipo en el futuro. En cuanto a los tests farmacológicos, aunque en el SB sí se considera la respuesta a fármacos bloqueadores de los canales del sodio entre los criterios diagnósticos, en el SQTL el test de epinefrina no está incluido. El test de epinefrina, validado en diferentes protocolos por Shimizu et al y por el grupo de la Clínica Mayo16,28,29, contribuye a aumentar la precisión diagnóstica de los hallazgos genéticos dudosos o no fácilmente interpretables, sobre todo en casos de SQTL tipo 1. Dichos trabajos señalan que el test de epinefrina aporta un elevado valor predictivo negativo (hasta el 96%) para descartar que el hallazgo genético sea casual y hace patente un fenotipo que se mostraba oculto en el ECG basal. En nuestra serie (fig. 2A), encontramos a sujetos portadores de mutación en SCN5A y KCNH2 con intervalo QTc normal. La realización del test de epinefrina en una de estas familias, pese a que la mutación no estaba en KCNQ1, sino en KCNH2, posibilitó el diagnóstico en 3 miembros afectados (fig. 4B). Como muestran nuestros resultados, el test farmacológico con epinefrina o flecainida se debe considerar no sólo en probandos con fenotipo negativo, sino también en familiares; en ocasiones, como en el caso del SB mostrado en la figura 4A, el caso índice muestra fenotipo negativo y no cumple criterios diagnósticos, con lo que el diagnóstico no se alcanza. Fue el estudio de los familiares mediante test con flecainida lo que permitió alcanzar el diagnóstico de SB en el probando que compartía la mutación con los portadores silentes. Sin embargo, a la vista de nuestros resultados, los hallazgos de los tests farmacológicos deben ser interpretados con precaución: que no aparezca alteración en respuesta a epinefrina o flecainida en un sujeto portador genético no implica ausencia de enfermedad, ya que el test de epinefrina es de difícil interpretación y tiene baja sensibilidad según el genotipo subyacente, y el test de flecainida puede ser de resultado intermitente en un mismo paciente30.
LimitacionesEl escaso número de casos no permite extrapolar las conclusiones de este estudio a poblaciones más amplias. Son necesarios estudios con mayor tamaño muestral y fundamentalmente de carácter multicéntrico. Otra limitación del presente estudio es el uso de flecainida en lugar de ajmalina para desenmascarar el SB, ya que es conocida la mayor sensibilidad de esta. Por último, el estudio genético, aunque convenientemente interpretado, no garantiza al 100% la presencia de la enfermedad en caso de hallarse una mutación no previamente descrita ni descarta la enfermedad en caso de no detectarse variaciones genéticas. Por ello, dado que se excluyeron casos con probable canalopatía por no tener un mutación en los genes secuenciados (KCNQ1, KCNH2 y SCN5A principalmente), no se puede descartar la presencia de mutaciones en otros genes como la rianodina o calsecuestrina en los sujetos con genotipo negativo e incluso en quienes se encontró una mutación probablemente patogénica. Por ello, consideramos que el diseño del estudio de tipo transversal, con utilización del estudio genético como prueba de referencia para el diagnóstico, tiene esta limitación de excluir casos con genotipo negativo en los que no se puede descartar la enfermedad.
CONCLUSIONESLos criterios diagnósticos actuales para canalopatías han tenido baja sensibilidad en nuestra muestra de pacientes y familiares portadores de mutación patogénica. Esto ha sido más acentuado en el grupo de familiares asintomáticos, que muestra una muy baja penetrancia, con alta prevalencia de ECG basal normal. En contextos sospechosos de posible canalopatía (FVI, muerte súbita inexplicada o síncope de perfil cardiogénico sin aparente cardiopatía), el uso combinado de los tests genéticos y farmacológicos en pacientes y familiares contribuye a aumentar el rendimiento del protocolo diagnóstico para detectar casos de SQTL y SB con fenotipo basal no diagnóstico.
CONFLICTO DE INTERESESNinguno.