Cardiovascular diseases, including cardiomyopathy, are the major complications in diabetes. A deeper understanding of the molecular mechanisms leading to cardiomyopathy is critical for developing novel therapies. We proposed phosphoinositide3-kinase gamma (PI3Kγ) as a molecular target against diabetic cardiomyopathy, given the role of PI3Kγ in cardiac remodeling to pressure overload. Given the availability of a pharmacological inhibitor of this molecular target GE21, we tested the validity of our hypothesis by inducing diabetes in mice with genetic ablation of PI3Kγ or knock-in for a catalytically inactive PI3Kγ.
MethodsMice were made diabetic by streptozotocin. Cardiac function was assessed by serial echocardiographic analyses, while fibrosis and inflammation were evaluated by histological analysis.
ResultsDiabetes induced cardiac dysfunction in wild-type mice. Systolic dysfunction was completely prevented, and diastolic dysfunction was partially blocked, in both PI3Kγ knock-out and kinase-dead mice. Cardiac dysfunction was similarly rescued by administration of the PI3Kγ inhibitor GE21 in a dose-dependent manner. These actions of genetic and pharmacological PI3Kγ inhibition were associated with a decrease in inflammation and fibrosis in diabetic hearts.
ConclusionsOur study demonstrates a fundamental role of PI3Kγ in diabetic cardiomyopathy in mice and the beneficial effect of pharmacological PI3Kγ inhibition, highlighting its potential as a promising strategy for clinical treatment of cardiac complications of diabetic patients.
Keywords
Diabetes mellitus (DM) is a major health problem, affecting almost 400 million people and killing 5 million people a year worldwide.1 Cardiovascular complications, including atherosclerosis and cardiomyopathy, are the major causes of fatalities in DM.
Several cardiac abnormalities are observed in DM, including apoptosis, hypertrophy, fibrosis, and alterations of cardiomyocyte contractility, leading to left ventricle dysfunction. These abnormalities lead to the clinical condition known as diabetic cardiomyopathy (DCM), which is diagnosed when ventricular dysfunction occurs in the absence of coronary atherosclerosis and hypertension. Diabetes mellitus has been demonstrated to increase the risk of heart failure by 2 to 5 times.2
Diabetic cardiomyopathy may have several causes, as different pathological changes induced by the hyperglycemic state, such as oxidative stress, endothelial dysfunction and altered inflammatory profile, may affect cardiac function and structure.3
A deeper understanding of the molecular mechanisms leading to pathological cardiac abnormalities is critical for the development of novel therapies aimed at ameliorating the cardiovascular outcome of diabetic patients.
We have extensively studied in both cardiac cells and leukocytes the signaling of the gamma isoform of the phosphoinositide3-kinase gamma (PI3Kγ), a lipid and protein kinase that generates the second messenger phosphatidylinositol (3,4,5)-trisphosphate, leading to activation of downstream signaling cascades. When we studied the role of this protein in cardiac remodeling in response to pressure overload, we found that in cardiomyocytes, PI3Kγ negatively controls cyclic adenosine monophosphate (cAMP) levels as a scaffold protein4,5 and plays a pivotal role during the internalization of activated β-adrenergic receptors via its kinase activity.6 Moreover, since it is known that PI3Kγ regulates leukocyte migration by triggering the accumulation of phosphatidyl(3,4,5)-triphosphate at the leading edge,7 we further explored the impact of this signaling pathway in the inflammatory process involved in cardiac remodeling in response to pressure overload. We found that leukocyte PI3Kγ crucially affects the recruitment of inflammatory cells and the fibrotic process that are typically observed in pressure overload, since its inhibition allows favorable cardiac remodeling.8 At the vascular level, we have described that mice lacking PI3Kγ are protected from hypertension induced by chronic administration of angiotensin II9 by a mechanism involving both oxidative stress and calcium handling, thus suggesting an additional role of PI3Kγ in the regulation of vascular tone. Recently, we have synthesized a novel small pharmacological inhibitor of PI3Kγ, which looks promising in the fight against inflammation-associated cardiovascular diseases.10
The hypothesis of this study was that the PI3Kγ signaling pathway could be important in regulating leukocyte migration and cardiac remodeling, even in other contexts of cardiovascular pathophysiology, such as the cardiac alterations induced by DM. Thus, we tested whether PI3Kγ plays a role in the development of cardiac dysfunction induced by DM, and whether pharmacological PI3Kγ inhibition could exert a therapeutic effect against this complication of DM.
METHODSAnimal ModelMale 8-weeks old C57Bl/6J, knock-out (KO) mice for p110γ, the catalytical subunit of PI3Kγ, or knock-in mice with catalytically inactive p110γ (kinase-dead [KD]), as well as their respective wild-type (WT) littermates, were used. Transgenic mice were generated as previously described.5 Mice were housed in a 12-hour light/dark cycle with food and water ad libitum. Experiments were performed according to European (EC Council Directive 2010/6) and Italian (D.Lgs. 116/92) guidelines on animal care and were approved by an institutional board.
Diabetes mellitus was induced by serial streptozotocin injections. Streptozotocin (Sigma-Aldrich, United States), was dissolved in 2.94% Na citrate buffer, pH 4.5, at a concentration of 7.5mg/mL. Mice were injected intraperitoneally with 50mg/kg/d streptozotocin for 5 days.
The development of DM was assessed by testing serum glucose levels after a 12-hour fast by a glucose colorimetric assay kit (Cayman Chemical, United States), according to the manufacturer's instructions.
The GE21 was dissolved in water and administered by gavage at doses of 5mg/kg/d or 50mg/kg/d, as indicated. Administration of GE21 was started 7 weeks after the first streptozotocin injection.
Echocardiographic analysisMice were subjected to serial echocardiographic analyses up to 15 weeks after streptozotocin injections.
Echocardiographic analysis was performed in mice anesthetized with isoflurane (5% in 1L/min oxygen for induction, and 1% for maintenance of anesthesia), using a Vevo 2100 device equipped with 15-40MHz linear-array transducer (Visualsonics, Canada). All echocardiographic images were acquired when the heart rate of mice was between 540 and 590 bpm. A left ventricular M-mode tracing was obtained using the 2-dimensional parasternal short axis imaging as a guide. End-diastolic interventricular septum and posterior wall thicknesses, and left ventricular internal diameter were measured. Relative wall thickness, ejection fraction and fractional shortening were calculated according to standard formulas.
Then, in apical 4-chamber view, a pulse wave Doppler analysis was performed at the level of the mitral valve to measure the blood flow peak (E) at mitral inflow, and a tissue Doppler was used to assess the peak (E⿿) at the lateral mitral annulus. It can be argued that the E/E⿿ ratio is not a perfect marker of diastolic function, which could be better estimated with invasive methods such as pressure-volume loops. However, E/E⿿ has been demonstrated to be the most useful noninvasive estimator of diastolic function,11 and we chose this method because it allows assessment of diastolic function over time in the same animal.
Finally, we performed strain analysis as previously described.12 Briefly, using speckle⿿tracking-based strain analysis of 2-dimensional gray scale echocardiographic images acquired from the parasternal long- and short-axis views, strain and strain rate were quantified in the longitudinal and radial axes. All images were acquired at a high frame rate (about 200 frames per second).
HistologyMice were anesthetized, thoracotomy was performed, and the heart was arrested in diastole (intracardiac injection of 100μL KCl 1M). Hearts were removed and postfixed overnight in phosphate buffered saline 4% paraformaldehyde and paraffin-embedded. In cardiac slices (4μm), PI3Kγ was assessed by staining with a specific antibody (1:100), produced as previously described,6,8 followed by antimouse secondary antibody (1:200, Jackson Immunoresearch). Sections were also stained with anti-CD68 (1:200; AbD Serotec, United Kingdom), followed by antirat secondary antibody (1:200, Jackson Immunoresearch, United States) to assess macrophage infiltration. Moreover, fibrosis was assessed on sections stained with Sirius red and cover-slipped with xylene based mounting media.
All images were captured using a DMI3000B Leica fluorescence/optical microscope (Leica Microsystems; Germany) and processed for quantitative analyses with the Leica Application Suite (LAS V3.3) Image Analysis.
Statistical AnalysisData are expressed as mean ± standard error of the mean. Data were analyzed by 1-way ANOVA (analysis of variance) or repeated measures ANOVA as appropriate, followed by the Bonferroni post hoc test, using a statistical software (SPSS 21.0, IBM). A 2-tailed P < .05 was considered as significant.
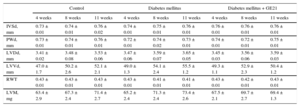
RESULTSEffects of Diabetes Mellitus on Physiological Parameters and Cardiac Structure and FunctionInfusion of streptozotocin-induced DM in 4 weeks raised fasting plasma levels of glucose from 180 ± 9mg/dL to 303 ± 30mg/dL (P < .05) and inhibited the increase in body weight induced by aging in control mice (24.4 ± 0.5g vs 26.1 ± 0.4g at the end of the observation period). The DM did not affect macroscopic indexes of cardiac structure. Similarly, PI3Kγ inhibition had no effects on the structure of the heart. In fact, neither measurements of thickness of interventricular septum and posterior wall nor diameter and volume of the left ventricle differed among the experimental groups (Table). Similarly, calculated parameters such as left ventricular mass and relative wall thickness were similar between diabetic and control mice (Table).
Ecochardiographic Measurements of Cardiac Structure
| Control | Diabetes mellitus | Diabetes mellitus + GE21 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| 4 weeks | 8 weeks | 11 weeks | 4 weeks | 8 weeks | 11 weeks | 4 weeks | 8 weeks | 11 weeks | |
| IVSd, mm | 0.73 ± 0.01 | 0.74 ± 0.01 | 0.76 ± 0.02 | 0.74 ± 0.01 | 0.75 ± 0.01 | 0.76 ± 0.01 | 0.76 ± 0.01 | 0.76 ± 0.01 | 0.76 ± 0.01 |
| PWd, mm | 0.73 ± 0.01 | 0.74 ± 0.01 | 0.76 ± 0.01 | 0.72 ± 0.01 | 0.74 ± 0.02 | 0.73 ± 0.01 | 0.74 ± 0.01 | 0.72 ± 0.01 | 0.75 ± 0.01 |
| LVDd, mm | 3.41 ± 0.02 | 3.48 ± 0.08 | 3.53 ± 0.06 | 3.47 ± 0.06 | 3.59 ± 0.07 | 3.65 ± 0.05 | 3.45 ± 0.03 | 3.56 ± 0.06 | 3.59 ± 0.03 |
| LVVd, mm | 47.0 ± 1.7 | 50.2 ± 2.6 | 52.1 ± 2.1 | 49.0 ± 1.3 | 54.1 ± 2.4 | 55.5 ± 1.2 | 49.3 ± 1.1 | 52.9 ± 2.3 | 50.4 ± 1.2 |
| RWT | 0.43 ± 0.01 | 0.43 ± 0.01 | 0.43 ± 0.01 | 0.43 ± 0.01 | 0.41 ± 0.01 | 0.41 ± 0.01 | 0.43 ± 0.01 | 0.42 ± 0.01 | 0.43 ± 0.01 |
| LVM, mg | 63.4 ± 2.9 | 67.3 ± 2.4 | 71.4 ± 2.7 | 65.2 ± 2.4 | 71.3 ± 2.4 | 73.4 ± 2.6 | 67.5 ± 2.1 | 69.7 ± 2.7 | 69.4 ± 1.3 |
IVSd, interventricular septum thickness in diastole; LVDd, left ventricular internal diameter in diastole; LVVd, left ventricular volume in diastole; LVM, left ventricular mass; PWd, left ventricular posterior wall thickness in diastole; RWT, relative wall thickness.
Ecochardiographic measurements of cardiac structure in control mice, mice treated with streptozotocin (50 mg/kg/day for 5 days), and mice treated with streptozotocin and the higher dose of GE21 (50 mg/kg/day, started 5 weeks after streptozotocin).
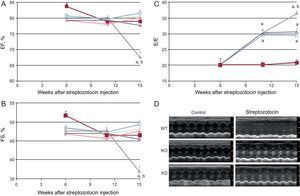
As expected, DM induced cardiomyopathy. In particular, an impairment of systolic function (evaluated as fractional shortening and ejection fraction, Figure 1) was observed 15 weeks after the induction of DM. Systolic function worsened further until the end of the observation period. Streptozotocin-induced DM also evoked diastolic dysfunction, evaluated as the E/E⿿ ratio. Diastolic dysfunction started from the 11th week after streptozotocin infusion and worsened further until the end of the study.

Genetic manipulation of PI3Kγ affects systolic and diastolic dysfunction in diabetic mice, as assessed by conventional echocardiography. A: ejection fraction. B: fractional shortening. C: E/E⿿ ratio of WT, KD and KO mice in control and diabetic conditions. D: representative echocardiographic images. EF, ejection fraction; FS, fractional shortening; KD, kinase-dead; KO, knock-out; PI3Kγ, phosphoinositide3-kinase gamma; WT, wild-type. WT mice, n = 11 per group; KD mice, n = 7 per group; KO mice, n = 4 per group. Full symbols, control mice; empty symbols, diabetic mice; ⿿, WT mice; , KD mice; ο, KO mice. aP < .01 vs control conditions. bP < .01 vs both KD and KO.
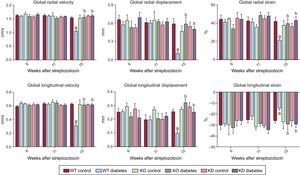
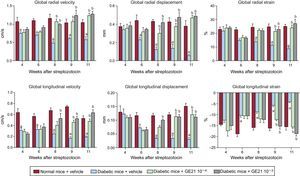
To assess cardiac function in a more detailed way, we performed strain analysis, observing that diabetic mice had a derangement of myocardial performance at the level of both radial and longitudinal motion (Figure 2). In fact, DM determined a decrease in both radial and longitudinal velocity displacement and strain. Similarly, DM reduced both radial and longitudinal strain.

Genetic manipulation of PI3Kγ affects cardiac dysfunction in diabetic mice, as assessed by strain analysis. Radial and longitudinal velocity, displacement and strain of WT, KD and KO mice in control and diabetic conditions. KD, kinase-dead; KO, knock-out; PI3Kγ, phosphoinositide3-kinase gamma; WT, wild-type. WT mice, n = 11 per group; KD mice, n = 7 per group; KO mice, n = 4 per group . aP < .01 vs control conditions. bP < .01 vs both KD and KO.
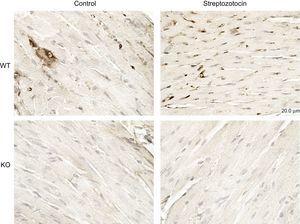
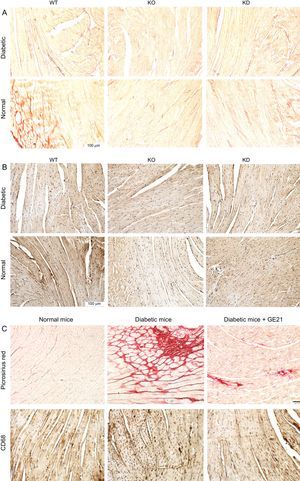
Diabetes mellitus was associated with the activation of PI3Kγ in the heart, as assessed by immunohistochemistry (Figure 3). Thus, we assessed whether PI3Kγ was involved in DCM by exploring mice genetically ablated for PI3Kγ (KO). Moreover, we tested whether these effects were due to the kinase activity of the enzyme by exploring mice expressing a KD form of the enzyme.

PI3Kγ expression is increased in diabetic mice. Representative microscopic images of histological preparations of left ventricles from control and diabetic mice. PI3Kγ KO mice were used as negative control. Magnification, ÿ200; scale bar, 100μm; n = 4 per group. KO, knock-out; PI3Kγ, phosphoinositide3-kinase gamma; WT, wild-type.
Neither genetic ablation nor the expression of a KD form of PI3Kγ affected fasting plasma glucose levels (KO, 288 ± 51mg/dL; KD, 295 ± 46mg/dL) or body weight, thus allowing us to look at the effect on DCM, independently of hyperglycemia. Moreover, in accordance with the data obtained in WT mice, DM did not affect macroscopic indexes of cardiac structure in any genotype (data not shown).
Basal cardiac function was similar in all genotypes. As stated above, DM induced systolic dysfunction in WT animals. The decrease in fractional shortening and ejection fraction, indexes of systolic dysfunction, was not observed in KO or KD mice (Figure 1). Similarly, the more detailed strain analysis showed that the cardiac dysfunction observed in WT mice 15 weeks after treatment with streptozotocin was not found in either KO or KD mice. In fact, longitudinal and radial velocity, displacement and strain were not affected by DM when PI3Kγ was ablated or mutated (Figure 2).
The role of PI3Kγ in DM-induced diastolic dysfunction had a different temporal profile. In the initial stages (up to 11 weeks after streptozotocin injection), both KO and KD mice showed the same decrease in E/E⿿ ratio as WT mice. However, while diastolic dysfunction continued to worsen in WT mice in subsequent weeks, diastolic function remained stable in mutant mice (Figure 1).
Thus, both the presence and kinase activity of PI3Kγ are important to mediate the development of cardiac dysfunction in response to DM.
Pharmacological Inhibition of PI3Kγ Counteracts the Systolic and Diastolic Dysfunction Observed in Diabetic MiceThese results prompted us to explore whether a pharmacological inhibitor of PI3Kγ kinase activity could revert DCM. For this aim, we treated mice with GE21 7 weeks after streptozotocin injection, ie, after DCM was already evident. In fact, cardiac dysfunction appeared earlier in this experimental series, as could be expected by the high variability usually associated with streptozotocin treatment.13
In addition, PI3Kγ inhibition did not affect fasting plasma glucose levels (337 ± 27mg/dL at maximal dose) or body weight (data not shown). Similarly, GE 21 did not affect macroscopic indexes of cardiac function (Table).
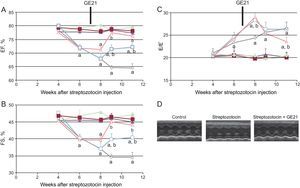
GE21 was able to counteract the decrease in fractional shortening and ejection fraction just 2 weeks after the beginning of treatment. The higher dose of this PI3Kγ inhibitor was even able to completely restore systolic function, making it similar to that observed in control mice. GE21 had no effects on systolic function in control mice (Figure 4).
. EF, ejection fraction; FS, fractional shortening. Full symbols, control mice; empty symbols diabetic mice; ⿿, treated with vehicle; , treated with 5 mg/kg/day GE21; ο, treated with 50 mg/kg/day GE21. aP < .01 vs control mice. bP < .01 vs vehicle-treated mice.")
GE21 reverts systolic and diastolic dysfunction in diabetic mice, as assessed by conventional echocardiography. A: ejection fraction; B: fractional shortening; C: E/E⿿ ratio of control and diabetic mice, treated with different concentrations of GE21; D: representative echocardiographic images (n = 10 per group). EF, ejection fraction; FS, fractional shortening. Full symbols, control mice; empty symbols diabetic mice; ⿿, treated with vehicle; , treated with 5 mg/kg/day GE21; ο, treated with 50 mg/kg/day GE21. aP < .01 vs control mice. bP < .01 vs vehicle-treated mice.
When we performed strain analysis, GE21 was able to counteract the decrease in both radial and longitudinal velocity, displacement and strain, providing diabetic mice with a complete recovery in myocardial performance (Figure 5). The effect of GE21 on radial velocity and displacement demonstrated an early effect of the drug, observable already after the first week of treatment. Moreover, the higher dose of the drug showed an earlier effect than the lower dose on longitudinal velocity and displacement. GE21 had no effects in control mice (data not shown).
. aP < .05 vs control mice. bP < .05 vs vehicle-treated mice.")
As observed with the genetic manipulation of the enzyme, the action of GE21 on diastolic function showed a different temporal profile of efficacy compared with that observed on systolic function (Figure 4). In fact, GE21 showed a biphasic effect: in the first weeks of treatment, GE21 further impaired diastolic function compared with untreated mice. This deleterious effect on diastolic function was brief, since it could not be observed from the second week of treatment. In contrast, after 4 weeks, GE21 treatment, at the higher dose of 50mg/kg/day started to counteract DM-induced diastolic dysfunction.
Both the early deleterious action and the late beneficial effect of GE21 on diastolic function depended on the interaction with mechanisms that were perturbed by DM, since GE21 had no effects on control mice.
PI3Kγ Modulates the Cardiac Fibrosis and Inflammation Observed in Diabetic MiceHistological analysis performed at the end of the experimental period showed that hearts from diabetic mice had a strong deposition of fibrotic material and macrophage infiltration (Figure 6).
. C: representative microscopic images of histological preparations of left ventricles from mice, treated with vehicle or 50mg/kg/d GE21, in control and diabetic conditions. Same staining as above (n = 10 per group; magnification, ÿ200; scale bar, 100 μm). KD, kinase-dead; KO, knock-out; PI3Kγ, phosphoinositide3-kinase gamma; WT, wild-type.")
Genetic and pharmacological inhibition of PI3Kγ reduces cardiac fibrosis and inflammation in diabetic mice, without affecting cardiomyocytes or vessels. A and B: representative microscopic images of histological preparations of left ventricles from WT, KD and KO mice in control and diabetic conditions (staining used in A is Picrosirius red, in B is anti-CD68; n = 4 per group). C: representative microscopic images of histological preparations of left ventricles from mice, treated with vehicle or 50mg/kg/d GE21, in control and diabetic conditions. Same staining as above (n = 10 per group; magnification, ÿ200; scale bar, 100 μm). KD, kinase-dead; KO, knock-out; PI3Kγ, phosphoinositide3-kinase gamma; WT, wild-type.
Both genetic inactivation of PI3Kγ activity and pharmacological inhibition of PI3Kγ, which were able to counteract DM-induced cardiac dysfunction, were also associated with lower fibrosis and macrophage infiltration.
DISCUSSIONDiabetic cardiomyopathy is a clinically distinct pathological entity of diabetic patients consisting of a cardiac dysfunction without concomitant risk factors such as obesity, hypertension, or smoking.14 Supposedly, 40% to 75% of diabetic patients have DCM,3 which increases the incidence of heart failure.15 Despite the high prevalence of this disease, there are currently no specific treatments for DCM patients. In this study, we observed that PI3Kγ play a fundamental role in the development of cardiac dysfunction in response to DM via its kinase activity, as observed in low dose streptozotocin-treated mice, a well-characterized model of slow-onset DM leading to gradual elevation of blood glucose levels.16 Accordingly, pharmacological inhibition of PI3Kγ can revert the cardiac dysfunction induced by DM. Finally, we found that this effect of PI3Kγ is associated to 2 important pathological determinants of DCM, ie, fibrosis and inflammation.
Our results show that the main effect of PI3Kγ inhibition in diabetics is an amelioration of cardiac function. In fact, DM caused evident cardiac abnormalities in both systolic and diastolic function. In contrast, no derangement of cardiac macroscopic structure was displayed during the entire observation period. These data are in accordance with previous reports,17 showing that streptozotocin-induced DM impairs cardiac function without modifying cardiac structure. The absence of PI3Kγ completely prevented systolic function even in the presence of DM. The same effect was observed with the genetic inhibition of PI3Kγ catalytic activity. On this issue, PI3Kγ exerts both kinase-dependent and kinase⿿independent effects on the heart.5 The fact that the simple genetic inhibition of catalytic activity is sufficient to obtain the same effects of the total absence of PI3Kγ indicates that PI3Kγ exerts its deleterious cardiac effects in DM only by its kinase-dependent action. The improvement of systolic dysfunction was demonstrated with greater sensitivity in strain analysis, which is a more detailed examination of cardiac function.
Several mechanisms can account for the cardiac effects of PI3Kγ in our study. PI3Kγ plays fundamental roles in leukocytes, cardiomyocytes, and vascular cells,18 all of which are involved in the cardiac response to DM.4 In particular, PI3Kγ has been demonstrated to directly control cardiomyocyte contractility through kinase-independent regulation of cAMP production.5 The fact that KD animals, which have cAMP levels comparable to WT,5 did not develop contractile dysfunction in response to DM rules out this possible explanation, although other possible direct effects in cardiomyocytes cannot be excluded. On the other hand, our study showed that the PI3Kγ genetic and pharmacological inhibition had a beneficial effect on 2 parameters affecting cardiac function in response to DM, ie, inflammation and fibrosis. The immune system participates in DCM development. In fact, mice models of DM show overexpression in the heart of cytokines such as interleukin-1β and transforming growth factor beta,19 and activation of macrophages.20 PI3Kγ is a fundamental mediator of immune system activation, and lack of its kinase activity inhibits leukocyte accumulation in the heart.8 Another pathological characteristic of DCM is induction of fibrosis. Increased perivascular and intermyofibrillar fibrosis has been observed both in human patients21 and in streptozotocin-induced mice.19 Moreover, these 2 actions are probably related, since activation of inflammatory pathways leads to increased fibrosis in mice with DCM.19 Accordingly, PI3Kγ has also been shown to induce cardiac fibrosis,5 and we have demonstrated that this feature is dependent on PI3Kγ in leukocytes,8 confirming the relevance of the link between cardiac inflammation and fibrosis in the cardiac action of PI3Kγ.
In this study, we also had a translational aim, by testing treatment with a pharmacological inhibitor as a novel strategy against DCM. A different pharmacological inhibitor of PI3Kγ prevented the development of autoimmune DM and reversed hyperglycemia in nonobese diabetic mice.22 This has raised expectation for anti-PI3Kγ treatment to fight the metabolic alterations observed in DM. However, it should be noted that the development of DM in nonobese diabetic mice has been ascribed to an autoimmune disorder.23 In autoimmune DM, pharmacological agents acting on the immune PI3Kγ pathway can be expected to effectively counteract the etiology of DM, more than its consequences. On the other hand, reversion of hyperglycemia was not observed in our study, in which DM was obtained through a direct toxic effect on pancreatic islet cells. Our study extends these previous observation on the effect of pharmacological inhibitors of PI3Kγ in DM, by showing that even in the absence of the metabolic effects that might be obtained in the fairly uncommon autoimmune DM, PI3Kγ inhibition still has a strong action against DCM.
In our study, genetic and pharmacological PI3Kγ inhibition counteracted DM-induced cardiac dysfunction. As observed with the genetic models, the main effect observed after treatment with GE21 was a strong improvement in systolic function, which was completely restored. The more detailed strain analysis showed an earlier effect of GE21 and also allowed a more accurate definition of the regional action of the drug. In fact, the amelioration of velocity and displacement on the radial axis of the left ventricle, which predominantly reflects the activity of the mesocardium (midmyocardium),24 was also observed with a lower dose of the drug earlier than corrections on the longitudinal axis, which instead mainly reflects the activity of the endocardium. However, the action on diastolic function was less clear. In fact, the initial development of diastolic dysfunction was not corrected by PI3Kγ ablation or inhibition of kinase activity, and only the subsequent progression of diastolic dysfunction was blocked. Similarly, with pharmacological inhibition, diastolic function was even impaired in diabetic mice, and only prolonged treatment with GE21 led to a rescue of DM-induced diastolic dysfunction. Our data suggest that 2 different mechanisms are responsible for the initial development of diastolic dysfunction and for its eventual progression in response to DM, and that PI3Kγ is involved only in the later stage. Therefore, chronic PI3Kγ inhibition seems to need a longer onset to achieve an improvement in diastolic features of the left ventricle. Since most DCM patients are characterized by prominent diastolic dysfunction,3,14 this could possibly limit the benefit of PI3Kγ inhibition in clinical practice. However, the overall significance of our results, showing long-term improvements in both systolic and diastolic function, suggests that PI3Kγ inhibitors could be considered as interesting potential treatments for the cardiac complications of diabetic patients. Future studies might indicate whether this line of investigation could lead to improvements in the clinical management of these patients.
CONCLUSIONSOur study demonstrates a beneficial effect of genetic and pharmacological PI3Kγ inhibition on DCM in mice. This study confirms the action of PI3Kγ inhibition on cardiac function, extending the validity of this approach to DM for the first time. The results of this study constitute a proof-of-concept highlighting the potential of a pharmacological approach based on PI3Kγ inhibition as a promising strategy for clinical treatment of patients suffering from DCM.
FUNDINGThis work was supported by Regione Campania (PON Distretto Campania Bioscience and POR-HOCKEY); and by Italian Ministry of Health (Ricerca Corrente) to G. Lembo.
CONFLICTS OF INTERESTNone declared.
WHAT DOES THIS STUDY ADD?- ⿿
We demonstrate a critical role for PI3Kγ in the development of DCM by using genetic models. Pharmacological therapy based on PI3Kγ inhibition was shown to be effective in a mouse model of DCM. Both genetic and pharmacological inhibition of PI3Kγ showed stronger activity against DM-induced decrease in systolic function than diastolic function.
We thank V. Berardi (IRCCS Neuromed) for assistance in the preparation and editing of the manuscript.