Bosentan has proven efficacy in pulmonary hypertension in the short term. Little is known about its effects beyond 2 to 3 years. Our objective was to analyze the efficacy and safety of bosentan in the long term (5 years) in patients treated in our center.
MethodsThis retrospective study sequentially analyzed clinical, functional, and laboratory parameters in a series of patients treated initially with bosentan as monotherapy from 2002 to 2009 in a single hospital. Treatment success was defined as survival without clinical worsening that required additional pulmonary vasodilators.
ResultsWe included 20 patients (70% women, mean age 46±14 years, 65% congenital heart disease), with a median follow-up of 64 months. One patient required withdrawal of bosentan due to adverse effects. At 4 months, significant improvements were achieved in hemodynamic, clinical and functional parameters. Clinical and functional benefits persisted at 5-year follow-up. Overall 5-year survival after beginning bosentan therapy was 95% (84%-100%). Treatment success at 1, 2, 3, 4 and 5 years was 95% (84%-100%), 83% (65%-100%), 78% (58%-98%), 61% (38%-84%), and 41% (16%-66%), respectively. The group with better outcomes had NT-proBNP levels at 1 year <400 pg ml P=.013).
ConclusionsIn our series, treatment success with bosentan in monotherapy was maintained in 78% at 3-year follow-up and 41% at 5-year follow-up. The group with long-term success showed significantly lower NT-proBNP levels at 1-year follow-up. Survival at 5 years in our series was 95%.
Keywords
Pulmonary hypertension is present in many clinical conditions and has been classified into 5 groups.1 Patients in group 1 —“pulmonary arterial hypertension” (PAH)— and group 3 —“chronic thromboembolic pulmonary hypertension” (CTEPH)— in which endarterectomy is not an option are dependant on pulmonary vasodilators, which improve patient survival and quality.2, 3
Endothelin-1, a peptide secreted by the pulmonary endothelium, promotes vasoconstriction and cell mitosis and increases pulmonary vascular resistance (PVR). Patients with PAH have higher concentrations of endothelin-1. Bosentan is a nonselective endothelin-A and endothelin-B receptor antagonist that leads to improved functional capacity and 6-minute walk test (6MWT) compared to placebo, as shown in short-term studies.4, 5 Clinical benefits have been demonstrated in specific groups of patients, such as those with idiopathic PAH,6 connective tissue disease,7 CTEPH,8 and congenital heart disease (CHD).9 Some advantages, shared with sildenafil, are oral administration and its relatively benign profile regarding adverse effects. For these reasons bosentan is used as first-line treatment in patients with moderately symptomatic PAH and initially those in functional class III, later extended to those in class II.10
Most of the studies on bosentan have had a mean follow-up of less than 1 year.4, 5, 7, 8, 9 Few studies have investigated whether its beneficial effects are maintained in the long term and none have followed patients for more than 3 years.11, 12, 13, 14, 15 This issue is of great relevance because these patients receive treatment for the rest of their lives.
The development of tolerance to the clinical benefits of pulmonary vasodilators has been reported. It has been shown that the benefits of beraprost, an orally active prostacyclin analog, obtained at 6 months are not maintained at 1-year follow-up, which has led to it being abandoned in our setting.16
The potential loss of effect of bosentan over the long term (>3 years), whether due to drug tolerance or to progression of the underlying disease, has not been investigated.
Our aim was to analyze the efficacy and safety of long-term (5 years) treatment with bosentan monotherapy in patients referred to our department and to identify those factors associated with persistent good response.
METHODS PopulationThe study included all patients diagnosed with severe PAH and CTEPH (Dana Point classification groups 1 and 3) in World Health Organization functional class II-III who started treatment with bosentan monotherapy between 2002 and 2009. Diagnosis was based on right heart catheterization, with a documented mean baseline pulmonary artery pressure (PAP) >45mmHg at rest. We excluded patients who started taking bosentan after treatment failure with other pulmonary vasodilators (3 patients) or who were taking bosentan and another pulmonary vasodilator simultaneously (4 patients).
Follow-up ProtocolFunctional class, 6MWT distance, and echocardiographic and laboratory parameters, including N-terminal fraction pro-brain natriuretic peptide (NT-proBNP), were recorded. Data were collected at baseline and every 6 months during treatment. Right heart catheterization was performed at baseline and after 3 months of treatment, and right atrial pressure, systolic PAP, diastolic PAP, mean PAP, pulmonary capillary pressure, cardiac output, cardiac index, and PVR were recorded.
All patients gave signed informed consent before starting treatment with bosentan. The starting dose was 62.5mg every 12h and after 4 weeks it was increased to 125mg every 12h. A monthly blood test was performed, including liver function parameters to monitor any adverse effects due to treatment.
EndpointsWe defined treatment failure as clinical worsening, usually manifesting as a worsening of functional class, the need for another pulmonary vasodilator due to poor symptom control, admissions related to PAH, transplantation, or death from any cause.
Statistical AnalysisNumerical variables are expressed as mean±standard deviation for normally distributed variables or as median (minimum-maximum) otherwise. Although functional class is an ordinal variable, it was also analyzed as a continuous variable because this made it possible to identify patients between the different classes and to better follow changes over time.
For normally distributed variables repeated measures ANOVA was used to compare repeated measurements over time, and nonparametric Friedman ANOVA was used for the remaining variables. Survival at final follow-up and treatment failure-free survival are expressed by Kaplan-Meier curves. In relation to survival analysis and other clinical data, we included data from all live patients to avoid the selection bias caused by only enrolling patients taking bosentan, regardless of whether changes in their treatment had been required or not. In this way, patient follow-up ended when death occurred, even in patients switched to combination therapy. On the other hand, for the actuarial analysis of treatment failure, patient follow-up ended when another drug was added.
To identify those variables associated with a good long-term response to bosentan monotherapy, we compared the baseline and changing characteristics of patients experiencing therapeutic success during follow-up to those who died or who experienced clinical worsening despite treatment. In addition, the characteristics of patients with CHD were compared to those with CTEPH.
In all cases, a P-value of <.05 was used as a cutoff for statistical significance.
RESULTS Baseline Characteristics of PatientsTable 1 shows the baseline characteristics of patients (n=20) included in the study.
Table 1. Baseline Characteristics of Patients (n=20).
| Age starting bosentan (years) | 46.2±14 |
| Women | 14 (70) |
| Clinical condition | |
| Systemic-to-pulmonary shunt | 13 (65) |
| Chronic pulmonary thromboembolism | 5 (25) |
| Systemic sclerosis | 1 (5) |
| Human immunodeficiency virus | 1 (5) |
| Concomitant treatment | |
| Home oxygen | 12 (60) |
| Oral anticoagulants | 17 (85) |
| Diuretics | 13 (65) |
| Calcium channel blockers | 3 (15) |
| Spironolactone | 6 (30) |
| Digitalis | 6 (30) |
| ACEI/ARA-II | 6 (30) |
| Phlebotomy | 2 (10) |
| Functional Class | |
| I | 0 |
| II | 3 (15) |
| III | 17 (85) |
| IV | 0 |
| Distance walked in 6min (m) | 314±66 |
| NT-proBNP (pg/mL) | 677.5 (26-4.734) |
| Echocardiographic parameters | |
| Pulmonary artery systolic pressure (mmHg) | 92±34 |
| Hemodynamic parameters | |
| Right atrial mean pressure (mmHg) | 9±4 |
| Pulmonary artery systolic pressure (mmHg) | 102±19 |
| Pulmonary artery mean pressure (mmHg) | 70.5±17 |
| Pulmonary capillary pressure (mmHg) | 12±4 |
| Transpulmonary gradient (mmHg) | 56±14 |
| Cardiac output (L/min) | 4.1±1 |
| Cardiac index (L/min/m2) | 2.5±1 |
| Pulmonary vascular resistance (WU) | 15±5 |
ACEI, angiotensin-converting enzyme inhibitors; ARA-II, angiotensin-II receptor antagonists.
Data are expressed as no (%), mean±standard deviation or median (minimum-maximum).
Congenital disease with systemic-to-pulmonary shunt was present in 13 cases (8 patients with ventricular septal defect, 3 with atrial septal defect, and 2 with persistent patent ductus arteriosus). Patients with PAH started bosentan monotherapy at a younger age than those in the CTEPH group (41.7±9 vs 62.8±13.3; P=.001).
The functional data (314±66min walked during the 6MWT) and hemodynamic parameters at baseline (PVR 15±5 WU) show that, in general, the study population presented advanced PAH and poor prognosis.
Baseline echocardiogram showed some degree of tricuspid regurgitation (80% in grades I and II) in all patients.
Treatment Efficacy ParametersOf the 20 study patients, treatment was discontinued in 1 patient after 1 month due to recurrent syncope. Median follow-up was 64 (13-96) months.
Short-term Effect (3-6 months): Clinical and Hemodynamic ParametersTable 2 shows how the clinical parameters evolved. Initial examination showed that 90% of patients were in functional class III. This parameter changed significantly after starting treatment with bosentan, and we recorded an improvement in functional class by 1 class in 74% of patients (14 moved from class III to II at 6-month assessment; 3 remained in functional class III and 2 remained in functional class II).
Table 2. Evolution of Other Parameters.
| Baseline | 6 months | 1 year | 2 years | 3 years | 4 years | 5 years | P | |
| Functional class | 2.8±0.3 | 2.1±0.3 | 1.9±0.4 | 2±0.5 | 2±0.5 | 2.2±0.4 | 2.3±0.5 | <.001 |
| 6MWT test (m) | 314±66 | 332±76 a | 363±76 | 361±73 | 360.7±72 | 351.2±94 | 351.6±124 | .003 |
| NT-proBNP (pg/mL) | 667 (26-4734) | 1.050 (43-4300) | 280 (25-4590) | 957 (69-7978) | 858 (98-9433) | 267 (88-3551) | 1.644 (87-5612) | .37 |
| PAP (mmHg) | 92±34 | 98±40 | 98±38 | 89±36 | 90±43 | 87.7±37 | 90±21 | .21 |
PAP, pulmonary arterial systolic pressure estimated by echocardiography; 6MWT, 6-minute walk test.
Data are expressed as mean±standard deviation or median (minimum-maximum).
a Not statistically significant compared to baseline.
With regard to hemodynamic parameters (Table 3), we documented a significant reduction in transpulmonary gradient and PVR, and increased cardiac output.
Table 3. Hemodynamic Parameters at Baseline and After 3 Months of Treatment.
| Hemodynamic Parameters (n=19) | Baseline | 3 months | P |
| Right atrial pressure (mmHg) | 9±4 | 9±8 | .9 |
| Systolic pulmonary artery pressure (mmHg) | 102±19 | 100±25 | .7 |
| Pulmonary artery pressure (mmHg) | 70.5±17 | 64±17 | .9 |
| Pulmonary capillary pressure (mmHg) | 12±4 | 14±7 | .2 |
| Transpulmonary gradient (mmHg) | 56±14 | 50±17 | .01 |
| Cardiac output (L/min) | 4.1±1 | 4.7±1 | .04 |
| Cardiac index (L/min/m2) | 2.5±1 | 2.8±1 | .08 |
| Pulmonary vascular resistance (UW) | 15±5 | 11±5 | .001 |
Data are expressed as mean±standard deviation.
Clinical and functional parameters:
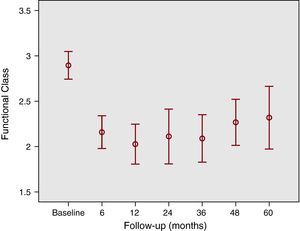
• The statistically significant short-term improvement was maintained during follow-up (figure 1). In numerical terms, the functional class was 2.8±0.3 at baseline, 1.9±0.4 at 1 year, 2±0.5 at 3 years and 2.3±0.5 at 5 years (P<.001 in relation to baseline in all cases).

Figure 1. Changes in functional class. Baseline functional class of patients (2.8±0.3) tends to significantly improve at 6 months and is maintained at 1, 2, 3, 4, and 5-year follow-up.
• 6MWT: we recorded a significant improvement in the distance walked during the 6MWT throughout follow-up. This increase was still evident after 1 year and remained significantly greater (P=.003) than at baseline in patients who were followed up at 2, 3, 4, and 5 years. The mean increase compared to baseline at 1, 2, 3, 4, and 5 years was 49, 48, 47, 38, and 38 m, respectively. Exercise capacity during the 6MWT was significantly greater in the PAH group than in the CTEPH group at 3 years (378±66 vs 278±31; P=.02) and 5 years (417±66 vs 220±111; P=.01).
• NT-proBNP: in our series, NT-proBNP values were dispersed over a wide range. There were no statistically significant differences at follow-up as compared to baseline values.

Echocardiographic parameters: all patients presented some degree of tricuspid regurgitation at baseline, which allowed systolic PAP to be estimated. Thus, the average value at baseline was 92±34mmHg, similar to that obtained by catheterization. There were no significant differences in this variable during follow-up despite clinical improvement in patients. Nor was there a significant change in the degree of tricuspid regurgitation over time.
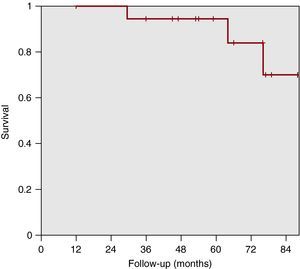
Overall Survival and Treatment Failure-Free Survival With BosentanAt a median follow-up of 64 months, there were 3 deaths, at 29, 64, and 75 months, respectively, 2 due to disease progression and 1 due to septic shock, although the patient was also in the terminal stage of the illness (Figure 2). These patients had previously presented clinical worsening that led to another vasodilator drug being added at 23, 41 and 53 months, respectively, to reestablish stability. Actuarial survival of patients was 95% (84%-100%) at 5 years and 70% (30%-100%) at 7 years.

Figure 2. Patient actuarial survival is 95% (84%-100%) at 5 years and 70% (30%-100%) at 7 years.
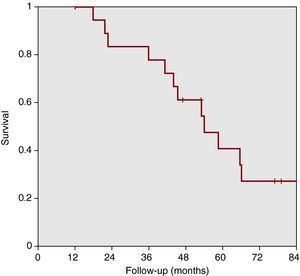
Figure 3 shows patient progress in relation to event-free survival or worsening requiring changes in treatment (failure of bosentan). The therapeutic success of bosentan monotherapy at 1, 2, 3, 4, and 5 years was 95% (84%-100%), 83% (65%-100%), 78% (58%-98%), 61% (38%-84%), and 41% (16%-66%), respectively.

Figure 3. Treatment failure-free survival. The therapeutic success of bosentan monotherapy at 1, 2, 3, 4, and 5 years was 95% (84%-100%), 83% (65%-100%), 78% (58%-98%), 61% (38%-84%), and 41% (16%-66%), respectively.
On average, treatment failure appeared in 12% of patients for each year of follow-up. In most cases, the pulmonary vasodilator administered with bosentan was sildenafil and this usually led to new clinical improvement, as shown by the benefits maintained in the clinical and functional parameters of the series.
Characteristics Associated With a Good Sustained Response to BosentanTable 4 shows the baseline characteristics and those after bosentan had been administered in the group of patients who experienced therapeutic success compared to those who died or underwent clinical worsening. There were no statistically significant differences between groups in relation to baseline characteristics. Neither were there differences between the groups in hemodynamic parameters as measured by catheterization at 3 months. Similarly, there were no changes in parameters (increased cardiac output and cardiac index, decreased PVR and mean PAP) at baseline and when measured by catheterization at 3 months.
Table 4. Characteristics of the Successful Treatment Group Compared to Those of the Failure Treatment Group.
| Parameter | Success (n=7) | Failure (n=12) | P |
| Baseline CO (L/min) | 4.4 (3.5-7.7) | 4.3 (3-7.3) | .7 |
| Baseline cardiac index (L/min/m2) | 2.4 (2.3-4.6) | 2.5 (1.9-4.2) | .4 |
| Baseline PVR (WU) | 11.4 (4-19) | 9 (6-21) | .6 |
| Increase in cardiac index (L/min/m2) | 0.76 (–0.3-1.9) | 0.1 (–0.9-0.5) | .3 |
| Increase in CO (L/min) | 0.59 (–0.4-3.3) | 0.6 (–1.5-1.8) | .7 |
| Reduction in baseline PVR (WU) | 5.6 (1.1-12.2) | 2.3 (–0.7-8.2) | .1 |
| Reduction mPAP (mmHg) | 13 (–27-29) | 5.5 (–20-20) | .1 |
| Baseline decrease of 1 FC at 6 months | 5 (71) | 9 (75) | .8 |
| Change in baseline FC at 12 months | 0.7±0.2 | 0.7±0.3 | .3 |
| Increased baseline 6MWT at 12 months (m) | 60.2±16 | 37.5±10.5 | .4 |
| 6MWT at 12 months (m) | 372±55.3 | 352±87 | .6 |
| 6MWT >350 m at 12 months | 4 (57) | 6 (50) | .7 |
| Increase of 50 m from baseline during 6MWT test at 12 months | 5 (71) | 5 (42) | .2 |
| NT-proBNP at 6 months (pg/mL) | 552 (122-1370) | 1530 (43-4300) | .3 |
| NT-proBNP at 12 months (pg/mL) | 210 (25-338) | 1431 (79-4590) | .04 |
| NT-proBNP <400 pg/mL at 12 months | 7 (100) | 4 (33) | .013 |
| Reduction of 200 pg/mL in NT-proBNP baseline at 12 months | 5 (71) | 3 (25) | .06 |
| AF during patient evolution | 1 (14) | 3 (25) | .5 |
AF, atrial fibrillation; CO, cardiac output; FC, functional class; mPAP, mean pulmonary arterial pressure; PVR, pulmonary vascular resistance; 6MWT, 6-minute walk test.
Data are expressed as no (%), mean±standard deviation or median (minimum-maximum).
Median NT-proBNP at 1 year after starting treatment with bosentan was significantly lower in the treatment success group: 210 pg/mL (25-338) vs 1431 pg/mL (79-4590) (P=.04). All patients who experienced therapeutic success had NT-proBNP <400 pg/mL at 1 year compared with only 33% in the group with worse outcomes (P=.013).
Thus, NT-proBNP values <400 pg/mL at 1 year after starting treatment with bosentan identified long-term responders with 100% sensitivity and 66% specificity, a negative predictive value of 100%, and a positive predictive value of 64%.
Safety and Adverse Effects (n=20)One patient (5%) developed recurrent syncope due to intolerance to bosentan leading to treatment being withdrawn 1 month after starting treatment. A transient increase of transaminases was observed in 3 patients during the first 4 months, but without requiring treatment withdrawal. No patient presented anemia or other laboratory abnormalities that required treatment. No significant adverse events were recorded.
DISCUSSIONThis study shows that there were significant improvements in clinical, functional, and hemodynamic parameters in the short term after treatment with bosentan in a series of patients with PAH due to various underlying clinical conditions. After a median follow-up of 5 years, clinical improvement and the distance walked during the 6MWT were both maintained. However, after a good response to treatment during the first year, approximately 12% of patients per year of follow-up presented clinical worsening, which in most cases required the addition of another pulmonary vasodilator.
In the short term, our experience replicates previous research regarding clinical, functional, and hemodynamic benefits. As reported in other studies,4, 5 clinical improvement of patients was more marked than improvement in hemodynamic parameters at 3 months of treatment. We observed significant but moderate improvements (range 10%-15%) in transpulmonary gradient and cardiac output, whereas PVR values, which show the cumulative benefit of both changes, showed a 27% reduction.
Compared with patients in other studies, our series had worse baseline PAH parameters. Patients included in other studies4, 5, 7, 8, 9 were able to walk >350 m at baseline during the 6MWT test, whereas the mean distance that our group could walk was 314 m. In addition, PVR in our group was 15 WU compared to 11 WU in Channick's et al. study.4
The present study differs from other studies on the effect of bosentan in PAH in that follow-up was far longer – more than double in some cases.
Studies with medium-term follow-up (between 1 year and 2.7 years) showed improvements in functional class, hemodynamic parameters, and distance walked during the 6MWT test in the first months of treatment, confirming the short-term outcome referred to above. The results obtained varied at 1-year follow-up. Diaz-Caraballo et al.15 obtained good results in a study that included 10 adults with CHD-associated PAH, as their functional class and 6MWT tests showed significant improvement at 25-month follow-up.
Three other studies showed less favorable results. The study by Provencher et al.,11 which included 103 patients with idiopathic PAH who were followed up for 2 years, showed treatment failure in 39% of patients after 1 year and in 56% after 2 years. In most patients, intravenous prostacyclin was added as rescue therapy. This worse outcome may be explained by the clinical condition underlying PAH (it is well known that CHD- associated PAH has a better outcome than idiopathic PAH), and by the fact that patients were in functional class III-IV at baseline.
The study by Van Loon et al.,14 which followed up 20 adults and 10 children with CHD for a median of 2.7 years, showed that bosentan significantly improved functional class and 6MWT test values at 4 months, and that the improvement continued up to 1 year. Subsequently, there was a decrease in benefit in the total series, which showed a 27% treatment failure at follow-up, largely in the pediatric population that had worse PAH at baseline (78% and 57% persistent benefit at 1-year and 2-year follow-up in adults and 50% and 20%, respectively, in the pediatric population). The worse prognosis of PAH in pediatric patients appears to be confirmed in a study by Apostolopoulou et al.13 This study included 19 patients, the majority of whom suffered from CHD-associated PAH and whose mean age was 22 years. Although no treatment failure was defined, in contrast to other studies their results show a loss of the initial clinical and functional benefit after 24 months of follow-up.
The favorable outcome of patients in our series (100% and 83% treatment failure-free survival at 1 year and 2 years, respectively) may be explained by the fact that the population studied consisted of adult patients with CHD-associated PAH. Although this outcome appears to be more favorable than that of other studies, it is similar to a recently published British series.17 Thus, the apparent discrepancy with the experience of other authors may in fact be less than it seems. It is important to note that in our group the longer the follow-up period, the greater the treatment failure (22% at 3 years and 59% at 5 years), suggesting that PAH progression eventually led to a slow clinical worsening of patients despite treatment.
It should be noted that the so-called therapeutic failure in these studies was not associated with a prognosis as unfavorable as the term might convey. In most cases, the addition of another specific drug (sildenafil or prostacyclin analogues) as rescue therapy changes the clinical outcome and patients recover the benefit of treatment, at least for a time, as shown in Hoeper's study18 and in our own experience.
In terms of mortality, it seems to be no significant differences compared to the few reports that have provided specific values. In the study by McLaughlin,12 which included 139 patients with idiopathic PAH in functional class III treated with bosentan, mortality was 3% at 1 year and 9% at 2 years, while in the study by Van Loon et al.,14 mortality was 10% at 2.7 years.
In comparison, there was 5% mortality at 5 years and 30% at 7 years in our series, which seems more optimistic in absolute terms and may be explained by differences in the underlying clinical condition and age of the study population. In any case, the values obtained are far from the mortality rates obtained in other series of patients receiving treatment with specific pulmonary vasodilators.3
The identification of a parameter associated with a prolonged and favorable response to treatment with bosentan is a useful aspect of this study. The values of the variable NT-proBNP were dispersed over a wide range and thus no significant differences were found for the whole group at follow-up despite the evidence of clinical improvement. However, when this variable was compared in the groups who were successfully treated and those in whom treatment failed at 5 years, we found that NT-proBNP values at 12 months after taking bosentan were significantly lower in the successful treatment group. In addition, 12 months after starting bosentan, all these patients had NT-proBNP <400 pg/mL. Thus, low levels of NT-proBNP may be associated with a good long-term response, with 100% sensitivity and 100% negative predictive value, which might provide a small but useful clue to the management of these patients. There is already widespread consensus that high levels of natriuretic peptides, and in particular their increase during follow-up, are independent predictors of mortality in patients with PAH.19 This fact must be taken with caution given the small size of our series. If confirmed in a larger series, this variable could identify a population at increased risk of death or clinical worsening and who should therefore undergo stricter follow-up and to whom lower thresholds should be applied for the adoption of other therapeutic measures.
Tolerance to bosentan was good, and treatment had to be withdrawn in only 1 patient (5%), a similar percentage to that reported by Van Loon et al.14 Three patients presented a transient asymptomatic transaminase increase, with values 3 to 5 times the upper normal threshold. This was managed with a temporary reduction of the dose and it did not reappear after bosentan was returned to the usual dose.
The limitations of our study are mainly related to the small size of the sample. However, medium-term series have in general a similar size (10-30 patients) and this is justified by the fact that PAH has a low prevalence. On the other hand, the predominance of CHD as the underlying clinical disease in our study may explain the high survival rate despite the unfavorable baseline parameters. In any case, the outcomes are similar to a recently published British series.17 Another limitation is the major alteration made in the echocardiography protocol during the study.20 Due to the lack of consistency in the determination of several echocardiographic parameters related to right ventricular function in our initial studies, we were not able to provide the same variety of echocardiographic data provided by other contemporary series of patients with PAH.
CONCLUSIONSOur experience suggests that bosentan monotherapy is a useful option to obtain short-term clinical improvement in patients with PAH. Improvement is expected to last for at least 1 year in most cases, with subsequent worsening in approximately 12% of patients per year and thus just under half of the patients receiving monotherapy would not undergo clinical worsening at 5 years. In other patients, characterized by high NT-proBNP values 1 year after starting treatment, the addition of other pulmonary vasodilators would usually be necessary to obtain further improvement. The 5-year survival rate in this series of patients was 95%.
FUNDINGDr Patricia Avellana has received funding via a research grant from the Fundación Carolina-BBVA. Drs. Javier Segovia, Manuel Gómez-Bueno, Pablo Garcia-Pavia and Luis Alonso-Pulpón belong to the Red Temática de Investigación en Insuficiencia Cardiaca REDINSCOR of the Instituto de Salud Carlos III, Ministerio Español de Ciencia e Innovación.
CONFLICTS OF INTERESTNone declared.
Received 23 January 2011
Accepted 25 April 2011
Corresponding author: Unidad de Insuficiencia Cardiaca, Trasplante e Hipertensión Pulmonar, Servicio de Cardiología, Hospital Universitario Puerta de Hierro de Majadahonda, Manuel de Falla 2, 28222 Majadahonda, Madrid, Spain. patriciaavellana@yahoo.com.ar