To the Editor:
The isolated form of non-compacted cardiomyopathy (NCC) is a rare disease whose underlying causes are increasingly well-understood due to greater knowledge and the development of imaging tools.
It is caused by the interruption of the embryonic process of myocardial compaction, which occurs between the fifth and eighth weeks of intrauterine life. As a result, the myocardium acquires a hypertrabeculated appearance with two layers, an external compacted epicardium and an inner, non-compacted endocardium, with deep intertrabecular recesses which communicate with the ventricular cavity.1,2
NCC frequently presents in families. In the series studied, up to 50% of family members are affected to some degree.
Mutations have been identified in several genes. The G 4.5, which is involved with mitochondrial function (taffazine protein), and others associated with the cytoskeleton (alpha-dystrobrevin and dystrophin) are inherited as sex-linked recessive genes. By contrast, genes that encode sarcomere proteins such as LDB3 (Cypher / Zaspa protein) on chromosome 10q21-q23, or cytoskeletal proteins such as alpha-actin or beta-myosin heavy chain, are inherited as autosomal dominant genes.3
Although NCC is classically expressed by the triad of heart failure, arrhythmias and stroke, our current capacity for early detection of incipient forms means that the clinical expression is often oligosymptomatic. It is associated with extracardiac, frequently neuromuscular, manifestations. However, the range is varied and includes facial dysmorphia, hematological disorders and endocrine or kidney disease, including horseshoe kidney, glomerulonephritis or polycystic kidney disease.2
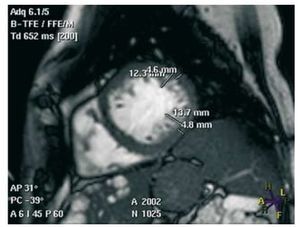
We report the case of 2 siblings with NCC and hepatorenal polycystosis. The index case was a hypertensive, dyslipemic woman aged 65 years who was on dialysis for renal failure secondary to polycystic disease. The woman was admitted with dyspnea at minimal effort, orthopnea, oliguria, and peripheral edema. The electrocardiogram sinus rhythm indicated left ventricular growth. An echocardiogram was requested and showed dilated left chambers with areas of distal hypertrabeculation. Contrast echocardiography in these areas indicated a mildly depressed ejection fraction and pulmonary pressure, compatible with "non-compaction cardiomyopathy." The diagnosis was confirmed by magnetic resonance imaging (Figure 1). After treatment with beta blockers and diuretics, the patient showed clinical improvements.

Figure 1. Short axis magnetic resonance showing the two layers of the myocardium, with a ratio of >2.3 in diastole.
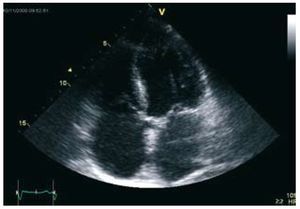
A review of the family history identified a 63- year-old brother who was also dyslipemic and hypertensive and who had received a kidney transplant for polycystic disease in 1995. Because of his history he had had several echocardiograms, all of which indicated dilation of the left-side cavities with moderate systolic dysfunction and apical hypertrabeculation compatible with NCC (Figure 2).

Figure 2. Apical four-chamber echocardiography showing hypertrabeculation in the middle segment of the anterolateral wall and apex, with a thickness 2 times greater than that of the compacted layer.
Polycystic kidney disease is a major cause of chronic kidney disease. It is hereditary and is produced by mutations which occur mainly in the PKD 1 and PKD 2 (polycystine 1 and 2) genes located on chromosomes 16 and 4 and which are transmitted as an autosomal dominant and recessive gene, respectively. Kidney cysts are associated with liver cysts. Signs of the disease include renal fossa pain, hematuria, and intercurrent urinary infections. Cardiovascular manifestations typically associated with the disease include hypertension, mitral valve prolapse, and intracranial aneurysms.
Only 3 cases of the association between NCC and polycystic kidney disease have been reported in the literature, and all reported the presence of both disorders in just one patient.4-6 We present the first concurrent presentation of both diseases in more than one case within the same family. Both siblings were diagnosed with hepatorenal polycystosis and cardiac abnormalities which met criteria for NCC.
Evidence exists of the genetic influence in these diseases. In the case of NCC, it appears that several causal genes remain to be identified. We do not know to what extent the association of these two conditions is a coincidence or the result of alterations in a single chromosome mechanism. Genetic studies of reported cases will undoubtedly help to resolve this issue.