Keywords
PATHOGENESIS OF THE ATHEROTHROMBOTIC PROCESS
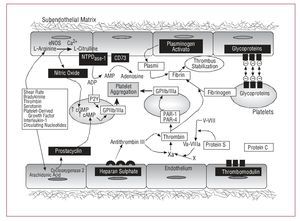
The endothelium is an autocrine and paracrine organ that regulates secretory, mitogenic, and contractile activities in the vascular wall and hemostatic processes through producing and releasing numerous active substances. In physiological conditions, endothelial cells exhibit multiple antithrombotic and fibrinolytic properties that prevent pathological thrombus formation (Figure 1).

Figure 1. Physiological antithrombotic mechanisms associated with the endothelium. cAMP indicates cyclic adenosine monophosphate; eNOS, endothelial nitric oxide synthetase; cGMP, cyclic guanosine monophosphate.
However, chronic and repeated exposure to cardiovascular risk factors (viral infections, immunocomplexes, hemodynamic stress, tobacco products, high cholesterol concentrations, eicosanoid enzymes released by platelets and leukocytes in inflammation, hyperhomocysteinemia, and diabetes) leads to activation/dysfunction of the endothelium characterized by the reduced bioavailability of nitric oxide and all the physiological cardiovascular protection mechanisms that derive from it.1 Such endothelial abnormalities facilitate the accumulation and oxidation of low-density lipoproteins (LDL) on the wall, which, being a potent inflammatory stimulus, leads to infiltration by circulating monocytes. Once in the subendothelial space, the monocytes differentiate into macrophages that take up great quantities of lipids and become foam cells (fatty streaks). Growth factors, cytokines, and other substances released by endothelial cells, monocytes/macrophages, platelets, and T-lymphocytes not only enable the development and deterioration of the endothelial lesion, but also potentiate the migration, proliferation, and synthesis of the extracellular matrix by the vascular smooth muscle cells. The result is a chronic inflammatory and fibroproliferative response that causes the development of atherosclerotic lesions. It is precisely the erosion or rupture of these atherosclerotic lesions that exposes prothrombotic surfaces leading to platelet adhesion, activation, and aggregation with the consequent thrombus formation (atherothrombosis). These thrombi not only contribute to asymptotic growth of atherosclerotic plaque, but are the leading cause of ischemic coronary syndromes.2
FACTORS DETERMINING THROMBUS FORMATION
Following vascular injury, the dynamics of platelet deposition, and the consequent formation of the thrombus are locally regulated by the following factors: a) the grade of stenosis; b) the type of lesion; and c) the composition of the atherosclerotic plaque.1
Grade of Stenosis and Local Flow
The coronary lesion that causes the infarction is, in general, slightly stenotic, indicating that the exposed surface is the determining factor of the acute occlusion, rather than the grade of stenosis of the lesion. However, the acceleration and later deceleration of blood when passing through a stenosis contribute to thrombus formation and its structural characteristics.3,4
Type of Lesion (Surface Exposed to the Bloodstream)
It is thought that most ischemic coronary syndromes are the result of a sudden luminal thrombosis arising from various pathological situations: a) erosion of the atherosclerotic lesion; b) plaque rupture; and c) calcified nodules.5 Our group previously demonstrated3,6 that the exposure of deendothelialized vascular wall (vascular erosion) to increased blood flow (similar to that of a stenotic coronary artery) gives rise to a mild thrombogenic stimulus that is usually limited to significant platelet deposition on the exposed vessel, reaching a peak 5-10 min after exposure, but which may later become embolized by the current flow thus causing a minimal and transitory occlusion. In contrast, plaque rupture, and the consequent exposure of collagen fibers, and the internal components of the plaque, not only leads to twice the amount of platelet deposition caused by erosion, but while still subject to high blood flow rates, the thrombus is not completely embolized and remains partially anchored to the damaged surface, and thus a relatively persistent thrombotic occlusion occurs. The characteristics of the vessel (eg, underlying lesion progression) exposed by erosion and rupture determine the type of thrombotic response. Furthermore, the presence of this mural thrombus acts as powerful thrombogenic stimulus.7 Third, the least common of all the lesions is characterized by calcified nodules being exposed to the bloodstream. These present an underlying calcified plaque with superimposed bony nodules resulting in a discontinuous fibrous layer that, in turn, is lacking in endothelial cells.
Several studies have been based on the relative incidence of rupture, erosion, or the presence of calcified nodules. Studies on sudden death8 have found that, of the deaths with known underlying etiology, 55%-60% are due to plaque rupture; 30%-35%, to plaque erosion; and only 2%-7%, to the presence of calcified nodules. In contrast, studies based on autopsies of hospitalized patients have shown that there is a lower incidence (20%-25%) of erosion in myocardial infarction.9 However, these incidences may vary; plaque rupture processes occur more often in men under 50 years with abnormal lipid profiles, whereas the erosion process is more frequent in patients who smoke and in premenopausal women.5
Lesion Composition
The nature of the exposed surface is a key factor in an unstable plaque rapidly progressing to an occlusive thrombus or persisting as a nonocclusive mural thrombus.10 Using a perfusion chamber,11,12 our group analyzed the relative contribution of the different components of human atherosclerotic plaque (fatty streaks, sclerotic plaques, fibrolipid plaques, atheromatous core, and hyperplastic plaques) in the formation of the acute thrombus. The results show that the atheromatous core is the most thrombogenic substrate and thus is the atherosclerotic plaque component involving the highest risk of thrombosis after spontaneous or induced rupture.
THROMBUS FORMATION
Platelet Adhesion and Activation
The fracture or rupture of coronary atherosclerotic plaque, either spontaneously or induced (percutaneous coronary intervention [PCI]), exposes components of the vascular matrix leading to platelet adhesion, activation, and aggregation (thrombus), which can lead to an ischemic coronary event.
The platelet adhesion process includes the transportation of the platelets, by diffusion, toward the reactive surface and the interaction of the platelet membrane receptors with their respective ligands in the structures of the damaged wall (Figure 2). Extracellular matrix components that interact with platelets include different types of collagens, von Willebrand factor (vWF), fibrinogen, fibronectin, and other adhesion proteins, such as laminin, vitronectin, fibulin, and thrombospondin.13 Even though fibrinogen and vitronectin are not synthesized by vascular wall cells, they can be regarded as substrates relevant to thrombogenesis, since they are immobilized in the extracellular matrix in lesion sites.13

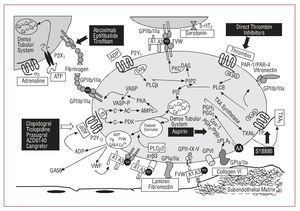
Figure 2. Main platelet activation and aggregation pathways and the resulting antiplatelet targets. AA indicates arachidonic acid; AC, adenylate cyclase; ADP, adenosine diphosphate; ATP, adenosine triphosphate; vWF, von Willebrand factor; PAR, protease-activated receptor; PG, prostaglandin; PI3K, phosphatidylinositol triphosphate; PKA, protein kinase type-A; PLC, phospholipase-C; TP, thromboxane receptor; TXA2, thromboxane A2.
Plasma vWF is basic to thrombus formation at high shear rates (>650/s), typical of small vessels, or partially occluded larger vessels. This is mainly due to the fact that high flow velocity conditions cause a change in the shape of vWF (from round to linear), leading to the possible exposure of the A3 domains of its multimers to vascular collagen (type I and type III collagen in the deepest vascular layers and type VI collagen in subendothelial layers) (Figure 2). On the one hand, such binding to collagen leads to a change in shape of vWF in its A1 domain that enables interaction with the glycoprotein (GP) Iba receptor of the platelet GPIb/IX/V complex and, on the other, enables interaction of the vWF Arg-Gly-Asp (RGD) domain with the platelet GPIIb/IIIa receptor (integrin aIIbb3).14 Therefore, we can see that circulating soluble vWF has to be immobilized in the arterial vessel to be able to interact with platelets.13 However, these 2 vWF-binding platelet receptors (GPIb/IX/V and GPIIb/IIIa) are promiscuous and thus bind to various ligands which in turn mediate adhesion to other platelets or cells. In this way, in the platelet adhesion and aggregation process, GPIIb/IIIa can bind with other key elements, such as fibrinogen, fibronectin, and CD40 ligand.13 Furthermore, the GPIb/IX/V complex can interact with P-selectin and the integrin leukocyte Mac-1 and perpetuate the inflammatory component of the atherosclerotic disease.
At low shear rates, the platelet adhesion function is mainly carried out by the collagen receptor that binds directly to platelet GPIa-IIa receptor (integrin a2b1).15 Fibronectin, laminin, vitronectin, and thrombospondin also contribute to this process, although to a lesser extent, by binding to GPIc-IIa and GPIIb-IIIa, GPIc'-IIa, vitronectin receptors, and GPIV and vitronectin receptors, respectively (Table).

In short, it can be considered that collagen and vWF are the primary agonists in the platelet activation and adhesion process, which is then promoted and enhanced by ADP released by the erythrocytes hemolyzed at the vascular lesion site, circulating thrombin and epinephrine, and the interaction of several agonists with their platelet receptors (Table). Most agonists that activate platelets act by hydrolyzing phosphatidylinositols in platelet membrane leading to the mobilization of free calcium in the dense tubular system of the platelet. This calcium release causes most of the platelet activation processes detailed below.
Exocytosis of the Content of the 3 Platelet Granules (Dense, Alpha, and Lysosomal)
The release of ADP, serotonin, and calcium (dense granules) favors the recruitment of new platelets, while the release of alpha granule growth factors initiates vascular repair, and the cytokines (alpha granules) and lysosomal enzymes (lysosomal granules) form the link to the immune response.
Eicosanoid Synthesis
Phospholipase A2 activates the arachidonic acid pathway; arachidonic acid becomes thromboxane A2 (TXA2) in a reaction that is catalyzed by the enzymes cyclooxygenase 1 (to form prostaglandin G2/H2) and thromboxane synthetase (to form TXA2).16 TXA2 is released into the circulation, where it binds to TXA receptors (TP receptors) on the adjacent blood platelet surface and amplifies the platelet activation process (Figure 2).
Adhesion Protein Expression
Translocation occurs at the surface of several adhesion proteins in the platelet alpha granules (P-selectin, GPIIb/IIIa, vWF, thrombospondin, fibrinogen, fibronectin, and vitronectin) responsible for the amplification and perpetuation of the platelet adhesion process, as well as the interaction of platelets with leukocytes.
Exposure of a Procoagulant Platelet Surface
A drastic change in platelet morphology (membrane) occurs, from a smooth disk to a spiny sphere, which promotes the exposure, in the outer membrane, of components of phospholipoprotein rich in phosphatidylserine. This facilitates the binding and activation of coagulation factors, as well as thrombin (a potent platelet agonist) formation.17
Platelet Aggregation
Regardless of the stimulus that leads to platelet activation, its final pathway is regulated by platelet GPIIb/IIIa receptor activation (Figure 2). The GPIIb/IIIa receptor is the most abundant protein on the platelet surface and consists of 2 protein subunits (IIb and IIIa). The activation of this receptor involves a change in the shape of the 2 subunits, such that the RGD binding domain is exposed to various ligands. Fibrinogen (of plasma or platelet origin) is the protein that mainly binds to the RGD domain of the GPIIb/IIIa receptor and its dimeric structure allows it to interact with 2 platelets at the same time, thus promoting platelet aggregation (Figure 2). Other GPIIb/IIIa ligands also participate, although to a lesser extent, in platelet-platelet interaction, such as vWF, fibronectin, vitronectin, and CD40 ligand.18
In the final stage of thrombus formation, fibrinogen is converted to fibrin by thrombin and stabilizes platelet aggregation. Thus, an expandable mass is formed that continues to recruit more platelets as these reach the prothrombotic microenvironment. Other blood cells are also recruited; erythrocytes and neutrophils are frequently found, and occasionally monocytes, that reach the lesion site intact, but respond to the presence of platelet secretion components.
In conclusion, atherothrombosis is, in most cases, the pathogenic event in acute coronary syndrome (ACS). Obviously, there are other possible, although infrequent, causes that can lead to coronary heart disease, such as dynamic obstruction of the vessel (coronary spasm or vasoconstriction), slow progressive mechanical obstruction of the vessel due to the growth of atherosclerotic plaque, or inflammation and infection processes. However, due to the essential role platelets play in the clinical symptoms, antiplatelet therapy, either aimed at inhibiting or blocking the signaling pathways and platelet activation, is the basic foundation for the treatment of coronary atherothrombotic disease (Figure 2).
ANTIPLATELET THERAPIES
Thromboxane Inhibitors
Aspirin
Aspirin (acetylsalicylic acid) causes the irreversible acetylation of cyclooxygenase (COX) in platelets (COX-1 isoform) and thus reduces TXA2synthesis (Figure 2).19 As platelets are anucleated, and are thus unable to carry out protein synthesis, they cannot replace the enzymatic activity, and thus platelet inhibition is maintained over the lifetime of the platelet (7-9 days). Aspirin does not inhibit platelet adhesion or aggregation in response to stimuli such as ADP, thrombin, collagen, catecholamines, and shearing stress. However, it has various non-platelet effects, such as prostaglandin inhibition, interleukin-6 synthesis inhibition in leukocytes, as well as reducing the activity of endothelial nitric oxide synthase (eNOS) inhibitors; this may help to explain why its beneficial effects are more than what would be expected of simple platelet inhibition based on a relatively weak agonist such as TXA2.
Although an initial 160-mg dose of aspirin is recommended to completely inhibit platelet Cox-1,20 the antithrombotic effect is, in general, relatively dose-independent, that is, lower quantities are able to almost completely inhibit TXA2 synthesis. Thus, the maintenance dose can be reduced to 81-100 mg/day for life. Furthermore, high doses of aspirin may cause serious adverse effects by inhibiting endothelial COX (COX-2 isoform), thus reducing the synthesis of PGI2 which is an important cardioprotector (Figure 1). We have to draw attention to the negative results obtained after selective inhibition of Cox-2.21
Regarding the role of aspirin in primary prevention, several metaanalyses have described the benefits of aspirin in patients with multiple risk factors (diabetes, stroke, peripheral arterial disease).20 Furthermore, in healthy middle-aged men, prophylactic aspirin can reduce the incidence of a first myocardial infarction by 44%.22 In contrast, there is a lack of data corroborating the efficacy of aspirin in primary prevention in women or the first appearance of stroke.23
Regarding secondary prevention, a large number of clinical trials have demonstrated the efficacy of aspirin in preventing ischemic disease (cardiovascular, cerebrovascular, and peripheral arterial disease) in high-risk patients.24 Thus, aspirin has become the reference antiplatelet agent and treatment with this should be initiated as soon as possible after the appearance of an acute event and be continued indefinitely, unless contraindicated by allergy, gastrointestinal complications, or hemorrhage.25 However, aspirin only reduces clinical events by approximately 30%. Furthermore, the increasingly common appearance of aspirin resistance and also the so-called "aspirin treatment failures" should lead to a deeper study of its effects and the development of new therapeutic alternatives. In fact, studies showing that aspirin is unable to protect against the onset of thrombotic complications, increases bleeding time, inhibits ex vivo platelet aggregation and inhibits platelet TXA2 production, have established that a high percentage (up to a barely believable 45%, depending on the study) of patients present "treatment failure" or "aspirin resistance," being more common in elderly people and women.26 The mechanisms involved in aspirin resistance are probably multifactorial and include the factors shown in Figure 3.

Figure 3. Possible mechanisms involved in aspirin resistance. NSAIDS indicates non-steroid antiinflammatories; CABG, coronary artery bypass surgery; EC, endothelial cells; COX, cyclooxygenase; TX, thromboxane.
To date, small studies have been conducted showing that incomplete thromboxane synthesis suppression, despite sufficient doses (aspirin resistance), is a potential cardiovascular risk marker.27 However, although the clinical implications of "aspirin resistance" are beginning to be understood, important issues remain unresolved, such as deciding what is the best diagnostic method to identify patients with "aspirin resistance" and what genetic factors and cellular mechanisms lead to "aspirin resistance." Similarly, clinical trials are needed to establish therapeutic guidelines for patients with "aspirin resistance" to reduce the risk of adverse events. Thus, various studies28 have pointed out that increasing the aspirin dose does not contribute any clinical benefit, but can in fact lead to a greater number of hemorrhagic complications.
Triflusal
Triflusal (2-acetoxy-4-trifluoromethyl benzoic acid)29 irreversibly inhibits platelet Cox-1 with almost no effect on endothelial Cox-2, thus preserving PGI2 synthesis. In addition to inhibiting thromboxane synthesis, it inhibits the action of phosphodiesterase. Although there are far fewer clinical trials have confirmed its efficacy, clinical benefits similar to those of aspirin have been demonstrated in patients after AMI or stroke events with a lower incidence of hemorrhage.30
Other Thromboxane Inhibitors
New thromboxane pathway inhibitors are currently being assessed, such as NCX-4016 (nitric oxide-releasing aspirin, an NO donor) and S18886, a thromboxane receptor antagonist. The advantages of NCX-4016 include all the benefits associated with aspirin plus the multiple cardioprotective properties of nitric oxide (antithrombotic, antiatherogenic, and vasodilatory properties). Numerous experimental animal studies have demonstrated the beneficial effects of NCX-4016 in the prevention of restenosis.31 S18886 is a reversible thromboxane (TP) receptor antagonist32 (Figure 2), and thus not only inhibits TXA2 activity, but also TP activation derived from other eicosanoids uninhibited by aspirin (HETE and isoprostanes).33 Our group has demonstrated the rapid and effective activity of S18886 in inhibiting stent-induced thrombosis without the concomitant increased risk of bleeding.34 Furthermore, it has been shown that S18886 prevents atherogenesis, causes regression of atherosclerotic plaque, and improves endothelial function in patients with coronary artery disease, and that these properties are relatively independent of the effects of TXA2 on platelets.35,36
Adenosine Diphosphate Receptor Antagonists
Ticlopidine and Clopidogrel
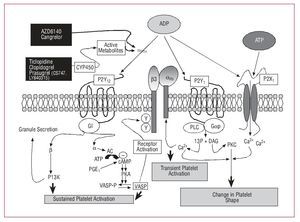
There are 3 receptors for ADP on the platelet surface, each of which initiates different platelet signaling pathways and different platelet aggregation functions; of the 3, P2Y12 is the main one in charge of sustained platelet aggregation (Figure 4).37

Figure 4. ADP platelet receptor signaling pathways. ADP indicates adenosine diphosphate; ATP, adenosine triphosphate; DAG, diacylglycerol; I3P, inositol triphosphate; PGE, prostaglandin E; PKC, protein kinase C; PLC, phospholipase C.
Much experimental evidence exists showing that of all the agonists released after platelet activation, ADP is one of the most important in the platelet recruitment and arterial thrombus propogation.37 Furthermore, numerous clinical trials have emphasized the importance of ADP, by demonstrating a reduction in ischemic events after treatment with ticlopidine38 and copidogrel.39
Ticlopidine and clopidogrel are antiplatelet agents derived from thienopyridines and inhibit APD-induced platelet aggregation, thus providing an effective alternative to aspirin when this is contraindicated (allergies, hemorrhages). Both are prodrugs and therefore have to be metabolized by the liver (by liver cytochrome P450 3A4, [CYP3A4]) to become active drugs with antiplatelet properties. The active metabolite covalently binds to cysteine residues of one of the ADP receptors (P2Y12), leading to irreversible modification of the receptor over the lifetime of the platelet.40
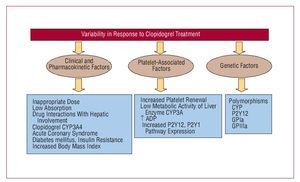
Ticlopidine was the first antiplatelet agent to be used from the thienopyridine family. Two important clinical trials, CATS38 and TASS,41 demonstrated the efficacy of ticlopidine in reducing thrombotic events in patients with atherosclerotic disease. However, antiplatelet activity began late and a maintenance dose of 250 mg twice a day only led to a 50% reduction in platelet aggregation at 5 days and a 60%-70% reduction at 8 days, thus limiting its use in patients with ACS. Furthermore, its regular use was significantly decreased by the incidence of neutropenia, although this was rare (8%thou). Thus, clopidogrel emerged as a new antiplatelet agent, similar to ticlopidine, but with fewer adverse effects (a 0.5%thou incidence of severe neutropenia). The milestone CAPRIE study confirmed the antiplatelet efficacy of clopidogrel versus aspirin. This study demonstrated that, in patients with history of myocardial infarction, stroke, or peripheral arterial disease, clopidogrel reduced ischemic event recurrence by 8.7% compared to aspirin.39 Furthermore, 2 h after administering a 600-mg loading dose, a significant reduction was observed in platelet aggregation, reaching its peak at 6 h (40%-60% reduction).42 In the light of these results, the hypothesis was put forward that a higher loading dose (900 mg) would accelerate the antithrombotic activity of the drug and overcome any limitation. However, the ALBION study43 demonstrated that a 900-mg loading dose did not lead to a faster onset of drug action, but did involve a greater tendency to hemorrhage. Another important limitation of clopidogrel, which remains unresolved, is the great interindividual variability observed regarding its antiaggregatory effect, the so-called "clopidogrel resistance."44 A certain number of patients have even been identified as "nonresponders" to clopidogrel who present an increased incidence of subacute thrombosis after stent implantation.45 Figure 5 shows the possible mechanisms that may be involved in this variability.

Figure 5. Possible mechanisms involved in the variability of response to clopidogrel.
The concept of "variability of response to clopidogrel" has led to concerns that some patients may not be adequately protected from platelet activation and aggregation and are thus exposed to a greater risk of thrombotic events.46 Pharmacological research has developed 3 new ADP receptor antagonists, prasugrel, AZD6140, and cangrelor, which are currently in phase II and III clinical trials and may expand the therapeutic arsenal available for these types of patients.
Finally, in addition to its antiaggregatory properties, it has been shown that clopidogrel reduces the formation of platelet-leukocyte conjugates in patients with ACS47 and reduces inflammation marker expression in activated platelets, such as CD40 ligand and the P-selectin, in patients undergoing PCI.48
Combined aspirin and clopidogrel therapy. The existence of complex interactions between the different platelet activation factors and the mechanisms involved in this process ensure redundancy in platelet activation pathways and thrombus formation. In clinical terms, this limits the efficacy of any individual agent and supports the use of combination therapy in various clinical settings. In patients with ACS, the CURE49 trial demonstrated the superiority of clopidogrel combined with aspirin versus monotherapy with aspirin, with a 20% reduction in relative risk. In fact, it is thought that it is the independent and complementary action of these 2 agents (both ADP-induced platelet aggregation and TXA2 production are inhibited) that provide the clinical benefits of this combination in ACS patients undergoing PCI (especially patients undergoing stent implantation).50 However, although the incidence of severe hemorrhage with aspirin in combination with clopidogrel and aspirin alone is the same, combination therapy is associated with a greater risk of hemorrhage. Thus, not all patients benefit from such dual antiplatelet therapy. In this context, this includes patients scheduled for PCI in urgent need of coronary artery bypass grafting (CABG).51 In fact, patients scheduled for CABG should not receive clopidogrel for at least 5 days before the intervention.52 Thus, many physicians are reluctant to prescribe clopidogrel until the coronary anatomy has been well defined. However, recent data from the CRUSADE53 study show that in the great majority of such patients CABG is performed within 5 days after the administration of clopidogrel, possibly based on the idea that the reduction of associated cardiovascular risk will counterbalance the increased risk of bleeding. Other clinical scenarios that also do not support the combined use of aspirin and clopidogrel include patients with cerebrovascular disease (increased bleeding)54 or stable cardiovascular disease with multiple risk factors. Recently, the CHARISMA55 study concluded that, compared to aspirin alone, combination therapy does not provide greater benefit in the primary prevention of vascular disease; these results call into question the concept of "the more, the better."
Prasugrel (CS-747, LY640315)
Prasugrel (CS-747, LY640315) is a thienopyridine, and thus requires previous metabolization by the liver to become an active drug that irreversibly inhibits platelet P2Y12 receptor.56 Compared to the standard dose of clopidogrel, the metabolization of prasugrel is more efficient (requiring a single metabolic step) and thus at lower doses more effectively inhibits platelet activity with less variability of response.57
The JUMBO-TIMI-2658 study assessed the safety of prasugrel versus clopidogrel in patients scheduled for PCI. The study concluded that, during the first 30 days after intervention, there were no significant differences between the 2 drugs regarding bleeding rates, but there were lower incidences of adverse cardiac events in patients treated with prasugrel, as well as lower incidences of myocardial infarction, recurrent ischemia, and venous thrombosis. The TRITON TIMI-3859 study directly compared the effects of prasugrel (60-mg loading dose and 10-mg maintenance dose) and clopidogrel (300-mg loading dose and 75-mg maintenance dose) on patients with ACS scheduled for PCI. Compared to clopidogrel, prasugrel was associated with a reduction in ischemic events, including stent thrombosis, although it has the drawback of higher bleeding rates, including fatal bleeding, in elderly patients, underweight patients, or those with stroke or transitory ischemic attacks.
AZD6140
AZD6140 is not a member of the thienopyridine family. It is a cyclopentyltriazolopyrimidine, and thus is orally active (does not require previous metabolization by the liver) acting directly and reversibly on P2Y12 receptor. These different pharmacodynamic and pharmacokinetic properties provide AZD6140 with possible advantages compared to thienopyridines, such as a faster and more powerful antiplatelet effect than clopidogrel.60
To date, the DISPERSE61 clinical trial, conducted with patients with atherosclerotic disease, confirmed treatment safety and reported more effective and faster platelet inhibition than that obtained with clopidogrel. The phase III clinical trial (PLATO)62 is underway, comparing the efficacy of AZD6140 with clopidogrel in patients with ACS.
Cangrelor
Cangrelor is also a reversible P2Y12 receptor inhibitor that is administered intravenously, unlike all the ADP receptor antagonists referred to. In patients with ACS (phase II trial),63 cangrelor has demonstrated its capacity for almost complete platelet inhibition and at a speed comparable to that of abciximab (GPIIb/IIIa inhibitor). Furthermore, compared to abciximab, platelet function was reestablished faster after stopping treatment, indicating a better safety profile. Currently, multicenter studies64,65 are being conducted to assess the clinical efficacy of cangrelor compared to clopidogrel in patients requiring PCI.
GPIIb/IIIa Antagonists
As mentioned, platelet activation occurs in response to various agonists that act via independent metabolic pathways (Table, Figure 2), but all of them converge toward a common final effect: GPIIb/IIIa (aIIbb3) activation and consequent platelet aggregation. This change in the shape of the receptor entails greater affinity for binding to soluble fibrinogen (through the RGD tripeptide sequence), as well as other adhesion molecules that also contain this RGD-binding domain (vWF, vitronectin, fibronectin). GPIIb/IIIa inhibitors are based on monoclonal antibodies that irreversibly block the fibrinogen binding site (abciximab) or small synthetic molecules that competitively and reversibly block the binding site for the RGD sequence (eptifibatide and tirofiban).66 The 3 drugs are administered intravenously, and large-scale clinical trials have demonstrated their clear clinical benefit and good safety profile in high-risk patients undergoing PCI.
Abciximab
Abciximab is made from the Fab fragment of the murine monoclonal antibody (7E3), subsequently chimerized to reduce its immunogenicity. Abciximab irreversibly binds to the GPIIb/IIIa receptor and blocks the binding of fibrinogen and other adhesion molecules that would lead to platelet aggregation. Unlike other GPIIb/IIIa antagonists, abciximab is not specific for this receptor and can also bind to vitronectin and leukocyte integrin Mac-1.67 This not only involves greater antiplatelet potential, but also confers a theoretical beneficial effect by reducing restenosis in patients undergoing PCI.68
Platelet inhibition occurs with abciximab in a dose-dependent manner. Following administration of a 0.25-mg/kg intravenous bolus, there is an almost complete reduction in platelet function, with more than 80% receptor occupancy. A continuous weight-adjusted infusion is required to maintain this degree of inhibition. The half-life of abciximab is long, between 6 h and 12 h. However, due to its irreversible binding with the receptor, the recovery of platelet function after stopping infusion is gradual and occurs 4-6 days after treatment. Thus, platelet transfusion is required to reverse its effects. Among the adverse effects of this drug, in addition to bleeding risk due to its great inhibitory potency, we would like to highlight its immunogenic potential—despite this being lower than that reported with murine antibodies—as well as thrombocytopenia (1.6%-5%).69
In clinical terms, abciximab has demonstrated its efficacy (up to 50% reduction of cardiac death or AMI) in patients with ACS requiring PCI, with and without later stent implantation (the EPIC, EPILOG, EPISTENT trials).70 In contrast, its clinical efficacy in patients with unstable angina not scheduled for PCI is less clear. Whereas several clinical trials (CAPTURE, PRISM, PRISM-PLUS, PURSUIT, TACTICS-TIMI 18, PARAGON A, PARAGON B)71,72 have demonstrated a clear benefit, differing results were obtained in the GUSTO IV study.73
In contrast to the benefits associated with intravenous administration of GPIIb/IIIa receptor antagonists, the results obtained by the administration of oral antagonists are discouraging. In fact, various large-scale clinical trials have demonstrated increased mortality after the administration of several oral preparations that, paradoxically, were associated with greater thrombosis.
Synthetic Molecules: Eptifibatide and Tirofiban
Eptifibatide and tirofiban are small synthetic molecules (and thus less immunogenic) that reversibly and competitively bind to fibrinogen receptor. Eptifibatide is a cyclic peptide based on the RGD sequence, whereas tirofiban is a nonpeptide derivative that mimics the RGD sequence. Both have a short half-life (2 h) and, due to the reversibility of their effect, their antithrombotic properties rapidly dissipate after stopping treatment.74
Various clinical trials have defended their efficacy in the treatment of ACS without PCI and in high-risk patients with diabetes mellitus or high troponin concentrations.75 However, in patients with ACS undergoing PCI, abciximab has demonstrated its superiority76 to these 2 agents (50% and 15%-35% reduction in events, respectively). It is thought that its greatest efficacy is due to the additional capacity of abciximab to block vitronectin receptor and leukocyte integrin Mac-1.
Thrombin Receptor Antagonists
Thrombin is not only an essential component in the coagulation cascade, but it is also fundamental to the atherothrombotic process.77 Thrombin activates the platelets by interacting with two G protein-coupled receptors (Figure 2) belonging to the protease-activated receptor (PAR) family, PAR1 and PAR4, and to a lesser extent, with GPIba (Table).78 PAR-1 has high affinity for thrombin and is thus the main agent in the signaling cascade triggered by thrombin-platelet interaction. PAR-4 has lower affinity and, together with GPIba, complements PAR-1 in the later phases of the activation process. Receptors are activated by a process of proteolysis, namely, thrombin binds to PAR cleaving the NH2-terminal domain and generates new NH2-terminal amino acid sequences (SFLLRN for PAR-1 and GYPGKF for PAR-4), which afterward stimulate other receptor sites.78 To date, clinical investigations have assessed two oral PAR-1 antagonists: E5555 and SCH530348. The efficacy and safety of E5555 are being assessed in patients with coronary arterial disease, while preliminary results79 in patients treated with aspirin and clopidogrel undergoing elective PCI indicate an excellent safety profile for treatment with SCH530348. The results obtained from phase III clinical trials (TRA-ACS and TRA 2P-TIMI-50) will eventually clarify the therapeutic potential of this new approach to antiplatelet agents.
FUTURE APPROACHES TO ANTIPLATELET AGENTS
Although there have been great advances in relation to the arsenal of antiplatelet agents for the treatment and prevention of ischemic heart disease, as presented in this review, these drugs still present certain limitations that leave certain groups of patients without protection. Thus, current efforts focus not only on enhancing the efficacy and safety of the agents presently in use, but also on developing new agents that, through blocking new targets, could become attractive therapeutic alternatives or be added to already established antiplatelet regimes without increasing the risk of bleeding. In this context, various experimental studies have demonstrated the efficacy of the administration of platelet-selective nitric oxide donors, platelet adhesion process inhibition, and Gas6 receptor blockade as possible therapeutic candidates.12,80-82 Nevertheless, we should continue to deepen our understanding of platelet activation pathophysiology. The great advances in genomics and proteomics are generating relevant information on the processes that integrate and link platelet agonist interactions, with the consequent activation of the signaling pathways and later thrombus stabilization, with the aim of developing new therapeutic targets that selectively inhibit the processes that trigger atherothrombotic disease.
This work was made possible due to financial support of PNS 2006 01091, CIBERobn-Instituto de Salud Carlos III, and Cátedra de Investigación Catalana-Occidente, UAB. GV has received a Juan de la Cierva postdoctoral research grant (Spanish Ministry Education and Science).
Correspondence: Prof.a L. Badimon.
Barcelona Cardiovascular Research Center.
Avda. Sant Antoni Maria Claret, 167. 08025 Barcelona. España.
E-mail: LBadimon@csic-iccc.org