
La ranolazina es un derivado piperazínico con un nuevo mecanismo de acción que ha sido aprobado para su uso en combinación en pacientes con angina crónica estable. La isquemia miocárdica aumenta la corriente tardía de entrada de Na+ en las células cardiacas (INaL) e incrementa la concentración de sodio intracelular ([Na+]i), lo que, a su vez, activa el modo inverso del intercambiador Na+-Ca2+ y aumenta la concentración de calcio intracelular ([Ca2+]i). Este aumento en Na+i y Ca2+i conduce a una disfunción mecánica (aumenta la presión diastólica y reduce la contractilidad y el aporte coronario de O2), eléctrica (produce arritmias) y mitocondrial (aumenta las demandas miocárdicas de O2 y reducción de la formación de adenosintrifosfato). La ranolazina inhibe selectivamente la INaL, reduce la acumulación intracelular de Na+ y la posterior de Ca2+ inducido por el Na+, así como las anomalías mecánicas, eléctricas y metabólicas en el miocardio isquémico o insuficiente. En ensayos clínicos controlados realizados en pacientes con angina crónica estable, la ranolazina ejerce acciones antianginosas y antiisquémicas y, en pacientes con síndrome coronario agudo, ejerce acciones antiarrítmicas. Además, reduce la glucohemoglobina en pacientes coronarios diabéticos y mejora la función ventricular en pacientes con cardiopatía isquémica o insuficiencia cardiaca crónica. La ranolazina es un fármaco bien tolerado cuyos efectos adversos más comunes son náuseas, mareos, astenia y estreñimiento. Por todo lo anterior, la ranolazina representa una alternativa segura y eficaz en pacientes con angina crónica estable con síntomas no controlados o que no toleran los fármacos antianginosos convencionales.
Palabras clave
A pesar de los indudables avances realizados en el diagnóstico y el tratamiento de la cardiopatía isquémica, ésta sigue siendo la primera causa de muerte en la Unión Europea (UE)1, con unos 740.000 fallecimientos anuales2. La angina crónica estable es la forma más frecuente de presentación de la cardiopatía isquémica en un 70% de los pacientes, con una incidencia anual del 0,5% en los mayores de 40 años, y conlleva una mortalidad anual de un 0,9-1,4%2,3. Según la Sociedad Europea de Cardiología, la prevalencia de angina aumenta en ambos sexos con la edad, desde un 0,1-1% en mujeres con edades comprendidas entre 45 y 54 años a un 10-15% en las de 65–74 años; en varones, estas cifras son de un 2-5% y un 10-20%, respectivamente. Por lo tanto, podemos calcular que en los países de la UE entre 20.000 y 40.000 individuos/ millón de habitantes presentan angina de pecho crónica estable2. Es importante señalar que estas incidencia y prevalencia siguen aumentando como consecuencia del envejecimiento de la población, el aumento de la arteriosclerosis y la mayor supervivencia tras infarto de miocardio. Finalmente, la cardiopatía isquémica se asocia a grandes morbilidad y costes, que en la UE se han estimado en unos 49 billones de euros anuales1,2.
El tratamiento convencional de la angina crónica estable se basa en la utilización de nitratos, bloqueadores beta, antagonistas del calcio y, en menor grado, otros fármacos (ivabradina, nicorandil y trimetazidina), y con frecuencia es necesario combinarlos a fin de controlar los síntomas del paciente4. Todos estos fármacos actúan: a) disminuyendo la demanda miocárdica de O2 (MVO2) como consecuencia de su capacidad para reducir la contractilidad y la frecuencia cardiacas, así como la tensión parietal (reducen la precarga y/o la poscarga), y/o b) aumentando el aporte coronario de O22,4 por su acción vasodilatadora. Sin embargo, un importante porcentaje de pacientes anginosos no toleran estos fármacos antianginosos o tienen comorbilidades (asma, hipertensión arterial, insuficiencia cardiaca, diabetes mellitus, bradicardia, bloqueo de la conducción auriculoventricular) que pueden contraindicar su uso o impiden alcanzar las dosis máximas recomendadas por el riesgo de que aparezcan cuadros de hipotensión arterial, depresión de la contractilidad cardiaca o bradicardia. Por otro lado, un porcentaje importante de pacientes con angina crónica estable siguen sintomáticos a pesar del tratamiento médico intensivo y de la utilización de técnicas de revascularización coronaria3,5,6. De hecho, al cabo de 1 año de haber realizado la revascularización coronaria, hasta un 25% de los pacientes siguen sintomáticos y un 60-80% requiere la administración de fármacos antianginosos5-7. De todo lo anterior se deduce que necesitamos nuevos fármacos antianginosos que actúen por mecanismos distintos, pero complementarios a los de los fármacos actualmente disponibles, que no presenten sus limitaciones (p. ej., que no modifiquen la frecuencia o la contractilidad cardiacas, la presión arterial o la conducción intracardiaca). Ello permitiría al combinarlos un mejor control de los síntomas del paciente.
RANOLAZINALa ranolazina [(+)-N-(2,6-dimetilfenil)-4(2-hidroxi- 3-(2-metoxifenoxi)-propil)-1-piperazina acetamida] (fig. 1) es un nuevo fármaco antianginoso y antiisquémico que presenta un mecanismo de acción novedoso, ya que es el primer fármaco que bloquea la corriente tardía de entrada de Na+ (INaL) en las células cardiacas8-12. Su uso ha sido aprobado en la UE para el tratamiento sintomático de pacientes con angina crónica estable, inadecuadamente controlados o intolerantes a los fármacos antianginosos clásicos13. En este artículo analizaremos, en primer lugar, el papel de la INaL en condiciones fisiopatológicas, en particular en el miocardio isquémico y, a continuación, el mecanismo de acción y las propiedades farmacológicas de la ranolazina.
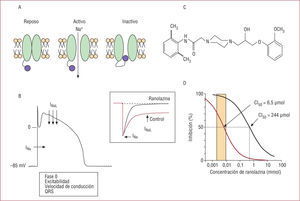
 y tardía (INaL). C: estructura química de la ranolazina. D: a concentraciones terapéuticas (≤ 10μmol), la ranolazina bloquea de forma selectiva la INaL en miocitos ventriculares obtenidos de un modelo canino de insuficiencia cardiaca, pero no modifica la INa. El recuadro muestra que la ranolazina no modifica la INa, pero inhibe marcadamante la INaL. CI50: concentración que inhibe en un 50% la amplitud de la corriente de Na+.")
A: cambios en la configuración del canal de Na+durante el potencial de acción cardiaco. B: potencial de acción cardiaco en el que se muestra cuándo tiene la activación de las corrientes de entrada de Na+rápida (INa) y tardía (INaL). C: estructura química de la ranolazina. D: a concentraciones terapéuticas (≤ 10μmol), la ranolazina bloquea de forma selectiva la INaL en miocitos ventriculares obtenidos de un modelo canino de insuficiencia cardiaca, pero no modifica la INa. El recuadro muestra que la ranolazina no modifica la INa, pero inhibe marcadamante la INaL. CI50: concentración que inhibe en un 50% la amplitud de la corriente de Na+.
En condiciones fisiológicas, la estimulación de las células musculares auriculares y ventriculares y de las células de Purkinje produce la activación-apertura de los canales de Na+, y se genera una corriente de entrada (INa) que despolariza el potencial de membrana y origina la fase 0 del potencial de acción cardiaco (y del complejo QRS en el ECG), así como la excitabilidad y la velocidad de conducción intraauricular e intraventricular (fig. 1B)14,15. Sin embargo, al cabo de 1–3ms, la mayoría de estos canales pasa al estado inactivo, que no permite que por ellos entre Na+al interior celular. Sin embargo, un pequeño porcentaje de canales de Na+ no se inactivan o se inactivan-cierran pero vuelven a abrirse de forma repetida durante la fase 2 del potencial de acción cardiaco, lo que genera una corriente tardía o lenta de entrada de Na+, que denominamos INaL. Esta corriente regula la duración de la fase 2 (meseta) y, por lo tanto, la duración del potencial de acción cardiaco16,17. Es decir, en los cardiomiocitos hay dos corrientes de entrada de Na+, una rápida que persiste unos pocos milisegundos y genera la fase 0, y otra tardía, que persiste unos cientos de milisegundos y participa en la repolarización y la duración del potencial de acción cardiaco. En condiciones normales, la amplitud de la INaL es sólo el 1% de la de la INa17 pero, dado que la INaL dura 50–100 veces más, la cantidad de Na+ transportada por la INaL es similar a la transportada por la INa.
Dos hallazgos confirman el importante papel que la INaL puede tener en la regulación de la actividad eléctrica cardiaca. El bloqueo de la INaL (p. ej., con lidocaína) acorta la duración del potencial de acción, mientras que los pacientes con síndrome de QT largo congénito Romano-Ward tipo 3 (SQTL3), que presentan mutaciones de los canales de Na+ que suprimen su inactivación, presentan un marcado aumento en la amplitud de la INaL en sus miocitos ventriculares, lo que sería la causa de la prolongación de la duración del potencial de acción (DPA) y del intervalo QTc del ECG18. La tabla 1 muestra que la INaL aumenta en diversas condiciones fisiopatológicas y se puede modular por mediadores endógenos y fármacos.
Situaciones patológicas y farmacológicas que aumentan o disminuyen la amplitud de la INaL
| Situaciones en que aumenta la amplitud de la INaLAdquiridasHipoxiaMetabolitos que se forman en el miocardio isquémico (amphiphiles)Radicales libres de oxígenoMediadores endógenos: trombina, angiotensina IIMiocardio insuficienteRemodelado ventricular tras infarto de miocardioHipertrofia ventricularMutaciones en los canales de Na+implicadas en el síndrome de QT largo (ΔKPQ)FarmacológicasToxinas: ATX-II, AP-A, β-PMTX, piretroidesFármacos: DPI 206–106, BDF 9148Segundos mensajeros: calmodulina, CaMKIIδInhibición de la INaLToxinas: tetradotoxina, saxitoxinaBloqueadores de los canales de Na+: mexitelina, lidocaína, flecainidaRanolazina |
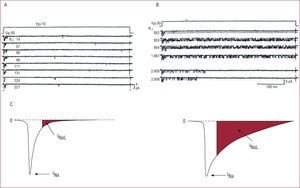
Kiyoshue et al16 demostraron por primera vez la existencia de ambas corrientes de entrada de Na+, INa e INaL en miocitos ventriculares de cobaya. En la figura 2 podemos ver que, al aplicar un pulso despolarizante, los canales de Na+ de los miocitos ventriculares de cobaya pueden presentar dos formas de apertura. En el panel de la izquierda observamos que los canales presentan una apertura muy corta al comienzo del pulso que genera la INa, seguida de alguna otra apertura ocasional que genera la INaL. En ocasiones (panel derecho), se observa que el canal de Na+ se abre y se cierra de forma repetida, lo que da lugar a la INaL. Hoy se piensa que ambas corrientes de entrada de Na+, INa e INaL las genera el mismo canal de Na+, si bien una porción de canales podrían presentar en un momento determinado una cinética de inactivación distinta, lo que podría estar relacionado con diferencias en la fosforilación del canal o la interacción de una proteína reguladora. A favor de que se trata del mismo canal está el hallazgo de que ambas corrientes de Na+ pueden generarse espontáneamente en células HEK293, en las que se expresa exclusivamente la subunidad alfa del canal humano de Na+(hNav1.5)19,20. El hallazgo21,22 de que los canales que generan ambas corrientes presentan la misma conductancia, el mismo tiempo de apertura y la misma selectividad iónica confirma que ambas corrientes se generan a través de una misma población de canales de Na+. Otra posibilidad es que la INa y la INaL pudieran generarse por distintas isoformas del canal de Na+ que se expresan de forma simultánea en los cardiomiocitos humanos, cada una de ellas con una cinética de inactivación distinta.
 de corta duración al comienzo del pulso que genera la INaL, seguida de alguna otra apertura ocasional que genera la INaL. B: en ocasiones se observa que el canal de Na+se abre y se cierra de forma repetida durante la aplicación del pulso, lo que da lugar a la INaL. C: representación esquemática de la magnitud de la INaL (en rojo) en ambas situaciones.")
Tras la aplicación de un pulso despolarizante, los canales de Na+de los miocitos ventriculares de cobaya pueden presentar dos formas de apertura16. A: aperturas (deflexión hacia abajo) de corta duración al comienzo del pulso que genera la INaL, seguida de alguna otra apertura ocasional que genera la INaL. B: en ocasiones se observa que el canal de Na+se abre y se cierra de forma repetida durante la aplicación del pulso, lo que da lugar a la INaL. C: representación esquemática de la magnitud de la INaL (en rojo) en ambas situaciones.
La isquemia cardiaca se caracteriza por un desequilibrio entre la demanda miocárdica y el aporte coronario de O2 que conduce a una alteración de la homeostasis iónica que, si es lo suficientemente marcada y prolongada, puede conducir a la necrosis de las células cardiacas23. Uno de los primeros hallazgos en la isquemia cardiaca es un aumento de la concentración intracelular de Na+ (Na+i)24-26. Este aumento podría tener lugar por tres mecanismos: a) la activación de la INaL: en situaciones de isquemia, la INa disminuye, mientras que la amplitud de la INaL aumenta marcadamente en respuesta a la hipoxia y a la generación de diversos metabolitos (p. ej., lisofosfatidilcolina) y los radicales libres aumentan la amplitud de la INaL11,19,27-31; b) durante la isquemia la glucolisis anaerobia y la degradación de la adenosina 5'-trifosfato (ATP) producen una rápida reducción del pH intracelular debido a la acumulación de ácido láctico y H+, lo que a su vez activa el intercambiador Na+-H+ (NHE); de esta forma se produce una entrada de Na+ como mecanismo compensador de la acidosis que contribuye al aumento de la Na+i32,33, y c) la inhibición de la ATPasa Na+-K+ producida como consecuencia de la reducción de las concentraciones celulares de ATP.
El aumento de la Na+i producido en los primeros momentos de la isquemia promueve, a su vez, la activación de la forma inversa del intercambiador Na+−Ca2+ (NCX, 3:1) que facilita la entrada de 1 Ca2+ en la célula y la salida de 3 Na+. Es decir, la activación del NCX a la larga contribuye, junto con otros mecanismos activados durante la isquemia (p. ej., la inhibición de diversas ATPasas de Ca2+), a que se produzca un aumento progresivo de la Ca2+i. Diversos hallazgos confirman que el aumento de la entrada de Na+ en las células cardiacas precede y causa la acumulación de Ca2+ en las células isquémicas: a) el aumento de la Na+i precede al aumento de la Ca2+i (fig. 2)26 ,34-37, y b) el aumento de la Ca2+i en el miocardio isquémico puede prevenirse bloqueando los canales de Na+25,36,38, inhibiendo el NHE32 o bloqueando el NCX39,40.
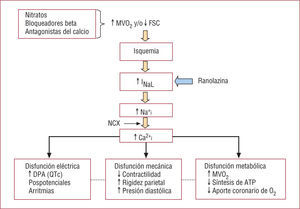
A su vez, el aumento de la Na+i y la Ca2+i en el miocardio produce varios efectos deletéreos (figs. 3 y 4)9-12,33,37,41
 y facilita la entrada de Ca2+ en la célula y aumenta la Ca2+i. El aumento de la Na+i y la Ca2+i produce alteraciones eléctricas, mecánicas y metabólicas. Esta secuencia puede inhibirse bloqueando la INaL con ranolazina. Los fármacos antianginosos clásicos actúan sobre los determinantes de la isquemia, mientras que la ranolazina actúa directamente sobre la isquemia. ATP: adenosintrifosfato; DPA: duración del potencial de acción cardiaco; MVO2: consumo miocárdico de O2.")
La isquemia produce un aumento de la INaL que conduce primero a un aumento de la Na+i, que a su vez activa al intercambiador Na+−Ca2+ (NCX) y facilita la entrada de Ca2+ en la célula y aumenta la Ca2+i. El aumento de la Na+i y la Ca2+i produce alteraciones eléctricas, mecánicas y metabólicas. Esta secuencia puede inhibirse bloqueando la INaL con ranolazina. Los fármacos antianginosos clásicos actúan sobre los determinantes de la isquemia, mientras que la ranolazina actúa directamente sobre la isquemia. ATP: adenosintrifosfato; DPA: duración del potencial de acción cardiaco; MVO2: consumo miocárdico de O2.
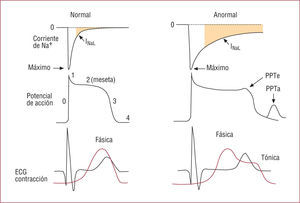
. Podemos ver que el aumento de la INaL (en rojo) inducido por la isquemia prolonga la duración de la fase de meseta (2) del potencial de acción (y del intervalo QTc), induce la aparición de pospotenciales tempranos (PPTe) y tardíos (PPTa) y una respuesta contráctil bifásica (fásica y tónica) que retrasa la relajación y aumenta la rigidez ventricular.")
INaL en condiciones normales y patológicas (cardiopatía isquémica o insuficiencia cardiaca). Podemos ver que el aumento de la INaL (en rojo) inducido por la isquemia prolonga la duración de la fase de meseta (2) del potencial de acción (y del intervalo QTc), induce la aparición de pospotenciales tempranos (PPTe) y tardíos (PPTa) y una respuesta contráctil bifásica (fásica y tónica) que retrasa la relajación y aumenta la rigidez ventricular.
Disfunción contráctil. El aumento persistente de la Ca2+i durante la diástole facilita la persistencia de los enlaces cruzados entre actina y miosina y retrasa la relajación cardiaca, lo que se traduce en un aumento de la rigidez y una menor distensibilidad de la cavidad ventricular durante la diástole que reduce la función ventricular (sistólica y diastólica) y conduce a un aumento del trabajo cardiaco, el consumo de ATP y la MVO29.
Disminución del aporte coronario de O2. Esta menor distensibilidad ventricular aumenta la tensión parietal, comprime los vasos coronarios intramurales y subendocárdicos y disminuye la perfusión coronaria a la zona isquémica durante la diástole5,33,37,42. Se cierra así un círculo vicioso por el que la propia isquemia acentúa los efectos deletéreos que ella misma produce.
Alteraciones de la función mitocondrial. La sobrecarga de Ca2+ inhibe aún más la síntesis mitocondrial de ATP. Puesto que la relajación cardiaca es un proceso activo que conlleva la activación de las ATPasas dependientes de Ca2+ del retículo sarcoplásmico y del sarcolema encargadas de reducir la Ca2+i, la reducción en la producción celular de ATP contribuye al aumento de la Ca2+i, el deterioro de la función diastólica y el aumento de tensión parietal y de la MVO2 en el miocardio isquémico.
Inestabilidad eléctrica. La INaL genera una corriente depolarizante que prolonga la DPA y el intervalo QTc del ECG. La amplitud de la INaL es mayor en las células M que en las del epicardio y el endocardio ventricular43,44. Como consecuencia, un aumento de la INaL prolongará mucho más la DPA en las células M o de Purkinje que en las musculares ventriculares, lo que se traduce en un incremento en la heterogeneidad de la repolarización ventricular y en una mayor dispersión del intervalo QT, dos marcadores de inestabilidad cardiaca que se asocian a la aparición de pospotenciales tempranos que pueden conducir a la génesis de arritmias cardiacas (fig. 4)9,45. Esta prolongación de la DPA conlleva una prolongación de la respuesta contráctil ventricular, de tal forma que, en vez de generarse una respuesta contráctil fásica, se genera una respuesta contráctil más prolongada (suma de una respuesta fásica y otra tónica) que retrasa la relajación ventricular, aumenta la rigidez de la pared ventricular y disminuye el aporte coronario de O2 durante la diástole23,46.
Por lo tanto, sería de esperar que el bloqueo de la INaL pudiera ser una alternativa terapéutica eficaz para reducir la Na+i y, de forma indirecta, la Ca2+i y para prevenir las alteraciones contráctiles, eléctricas y metabólicas que acompañan al proceso isquémico cardiaco41,42,47.
La ranolazina inhibe la INaLA concentraciones terapéuticas (≤ 10–21μmol/l), la ranolazina produce un bloqueo de la INaL dependiente de concentración, voltaje y frecuencia (CI50=5-21μmol/l) (fig. 1D)45,47,48. Este efecto es más marcado en miocitos cardiacos isquémicos tratados con peróxido de hidrógeno o con ATX-II (una toxina de Anemona sulcata que aumenta marcadamente la INaL) o procedentes de animales con insuficiencia cardiaca, ya que en todas estas situaciones la amplitud de la INaL está patológicamente incrementada (tabla 1)8,22,48-53. Sin embargo, a concentraciones terapéuticas, la ranolazina no modifica la INa encargada de la fase 0 del potencial de acción (IC50=244–294μmol/l)9-12,42,47-49,54-56, la corriente de entrada de Ca2+ (ICa) o la actividad de los intercambiadores NCX y NHE46,55,56. Ello explica por qué la ranolazina no modifica la contractilidad y la frecuencia cardiacas, la conducción intraauricular o intraventricular, las resistencias vasculares periféricas o la presión arterial.
Como consecuencia del bloqueo de la INaL, la ranolazina disminuye la Na+i, inhibe la actividad del intercambiador Na+-Ca2+ y la Ca2+i46,52,55,56, preserva la homeostasis iónica intracardiaca, reduce la tensión de la pared ventricular y mejora la distensibilidad y la relajación ventricular, a la vez que disminuye las MVO2 y aumenta el flujo sanguíneo coronario subendocárdico en modelos animales de isquemia-reperfusión (fig. 3). Es importante señalar que, a diferencia de los antianginosos clásicos, todos estos efectos se producen sin cambios en la presión arterial, la frecuencia cardiaca o la conducción auriculoventricular8,57,58. Por lo tanto, la ranolazina presenta un mecanismo antianginoso nuevo y complementario al de nitratos, bloqueadores beta y antagonistas del calcio que justifica su combinación en los pacientes con angina crónica estable, en particular, aquellos con bradicardia o presión arterial baja, para quienes los antianginosos clásicos están contraindicados.
Efectos antiisquémicos de la ranolazinaEn modelos experimentalesEn modelos de isquemia-reperfusión realizados en corazones aislados y perfundidos, la ranolazina tiene efectos cardioprotectores: preserva la concentración celular de ATP, reduce la liberación de creatincinasa y lactato deshidrogenasa, aumenta la velocidad máxima de aumento de la presión intraventricular izquierda (+dP/dt) y previene la acumulación de Ca2+ durante la reperfusión46,59. Puesto que estos efectos aparecen a concentraciones de ranolazina que no modifican los intercambiadores NCX y NHE o la ICa47,56, las acciones antiisquémicas de la ranolazina son consecuencia directa del bloqueo de la INaL. Por otro lado, dado que la ranolazina no modifica el tono vascular coronario, el aumento de la perfusión coronaria observada en estos modelos sería la consecuencia de la reducción de la acumulación de Ca2+ y la mejoría de la relajación ventricular durante la diástole46. En un modelo de miocardio aturdido (stunning), los animales pretratados con ranolazina presentaban un incremento de la fracción de eyección, mejoría de la función ventricular y disminución de los segmentos acinéticos/discinéticos46. Estos efectos eran independientes de cambios de la frecuencia cardiaca o de la presión arterial, que no se modificaban por el fármaco.
En ensayos clínicos controladosLos efectos de la ranolazina, en monoterapia o combinada con antianginosos convencionales, se han analizado en cuatro ensayos clínicos, tres realizados en 1.579 pacientes con angina crónica estable (MARISA [Monotherapy Assessment of Ranolazine in Stable Angina]60, CARISA [Combination Assessment of Ranolazine in Stable Angina]61, y ERICA [Efficacy of Ranolazine in Chronic Angina]62), y otro realizado en 6.560 pacientes con síndrome coronario agudo sin elevación del segmento ST (MERLIN-TIMI 36: Metabolic Efficiency with Ranolazine for Less Ischemia in Non-ST-Elevation Acture Coronary Syndromes- Thrombolysis in Myocardial Infarction 36)63,64. Tanto en monoterapia como combinada con antianginosos convencionales, la ranolazina es superior a placebo para reducir el número de ataques y el consumo de nitroglicerina, aumentar la tolerancia al ejercicio y prolongar los tiempos hasta la aparición de la angina o alcanzar una depresión de 1mm en el segmento ST60,61,63-65. En el estudio MERLIN-TIMI 36, la ranolazina no modificó la mortalidad total, pero mejoró la tolerancia al ejercicio y redujo la isquemia recurrente, el empeoramiento de la angina y la necesidad de utilización de fármacos antianginosos, y esta población en alto riesgo la toleró muy bien.
Los efectos antianginosos de la ranolazina aparecen a dosis del fármaco que no modifican la frecuencia o la contractilidad cardiacas, el flujo sanguíneo coronario, la presión arterial del paciente o el doble producto (frecuencia×presión), un índice del trabajo cardiaco57,58,60-62. Es decir, que su efecto antianginoso es diferente del de los fármacos antianginosos clásicos que reducen la frecuencia/contractilidad y las MVO2 y/o aumentan el flujo sanguíneo coronario57,58,60-62,66,67. Por ello, la ranolazina es una interesante alternativa terapéutica para pacientes que no se controlan con los antianginosos clásicos, así como pacientes con reducción de la presión arterial o de la frecuencia cardiaca, alteraciones de la conducción auriculoventricular o depresión de la contractilidad cardiaca, a los que no se puede administrar los antianginosos clásicos a las dosis indicadas o estos fármacos están contraindicados.
Efectos en la función ventricularUn importante porcentaje de pacientes con angina crónica estable presentan insuficiencia cardiaca (un 17-52% en los estudios clínicos realizados con ranolazina)60-62. En los cardiomiocitos de pacientes con insuficiencia cardiaca, se observa un aumento patológico de la INaL posiblemente relacionado con un incremento en la fosforilación de los canales de Na+ a través de la activación de la calmodulincinasa dependiente de Ca2+ tipo II (CaMKII)48,55,68. Este aumento de la INaL explicaría, en parte, las alteraciones contráctiles y eléctricas en el miocardio insuficiente8-12. La ranolazina inhibe la INaL y, por lo tanto, podría prevenir la acumulación patológica de Na+ y Ca2+, mejorar la función ventricular y prevenir las arritmias que aparecen en el miocardio insuficiente41.
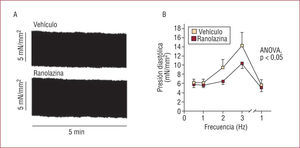
La ranolazina no modifica la contractilidad cardiaca en el miocardio normal8. Sin embargo, en miocitos aislados de perros o de pacientes con insuficiencia cardiaca, la ranolazina mejora la contracción y previene el aumento de la presión basal que aparece al estimular a frecuencias rápidas (fig. 5)48,51. En corazones aislados y perfundidos en los que se incrementaba la INaL por distintos métodos (isquemia-reperfusión, perfusión con ATX-II o con peróxido de hidrógeno), la ranolazina redujo el aumento de la Ca2+i durante la isquemia-reperfusión, disminuyó la presión telediastólica del ventrículo izquierdo y mejoró la recuperación de la función ventricular, pero no modificó el aumento de la Ca2+i (Ca2+ transient) o el flujo coronario46,50,69. También se ha observado que la ranolazina mejora la función ventricular (aumenta la+dP/dtmáx, el volumen latido y la fracción de eyección y disminuye la presión telediastólica del ventrículo izquierdo) en modelos experimentales de isquemia cardiaca47 y de insuficiencia cardiaca70,71. De nuevo, estos efectos se observaron en ausencia de cambios en la frecuencia cardiaca, la presión arterial, el flujo coronario o las MVO271.
, pero reduce el aumento de la presión diastólica basal (B), al estimular a frecuencias rápidas (3Hz) trabéculas obtenidas de ventrículos humanos de pacientes con insuficiencia cardiaca. Tomado de Sosalla et al51.")
La ranolazina no modifica la contractilidad cardiaca (A), pero reduce el aumento de la presión diastólica basal (B), al estimular a frecuencias rápidas (3Hz) trabéculas obtenidas de ventrículos humanos de pacientes con insuficiencia cardiaca. Tomado de Sosalla et al51.
En pacientes con disfunción diastólica ventricular tras infarto de miocardio (fracción de eyección media, 35%), la ranolazina aumenta la velocidad máxima de llenado ventricular izquierdo y la relajación segmentaria durante la fase de relajación isovolumétrica en los segmentos isquémicos, efectos que indican que el fármaco produce una mejora de la función diastólica72. Resultados similares se habían observado con una formulación de liberación inmediata de ranolazina a finales de los años ochenta en pacientes con cardiopatía isquémica y disfunción ventricular66.
Otros efectos farmacológicosEfectos electrofisiológicosLa ranolazina a concentraciones terapéuticas también inhibe el componente rápido de la corriente de salida de K+, que presenta rectificación interna (IKr, CI50=11,4μmol)45. El bloqueo de la IKr debería prolongar la DPA (y el intervalo QT del ECG) y podría facilitar la aparición de pospotenciales tempranos y arritmias ventriculares. Sin embargo, el desarrollo de torsades de pointes requiere tres condiciones47,56: la prolongación de la DPA y del intervalo QTc, la generación de pospotenciales tempranos y el aumento de la dispersión de la repolarización ventricular que crea las condiciones idóneas para la aparición de la reentrada del impulso cardiaco.
De hecho, en modelos animales, la ranolazina no aumenta la DPA en las células M y las fibras de Purkinje, que son las que generan pospotenciales tardíos arritmogénicos45,55, y a diferencia de los bloqueadores puros de la IKr-HERG, la ranolazina no prolonga la DPA o el intervalo QT a frecuencias cardiacas lentas52. En ventrículos de perro perfundidos, la ranolazina prolonga la DPA a nivel epicárdico y endocárdico (donde la INaL presenta menor amplitud y apenas si modifica el potencial de acción a nivel de las fibras de Purkinje, lo que se traduce en una reducción en la dispersión trans- mural de la repolarización)48,50,52,53,55. Además, en animales tratados con fármacos que bloquean la IKr (p. ej., d-sotalol, E-4031), la ranolazina no aumenta sino que acorta la DPA ventricular y el intervalo QT del ECG, reduce la dispersión de la DPA ventricular y suprime los pospotenciales tempranos y la incidencia de arritmias ventriculares8,48,50,52,53,55,62,73. Igualmente, suprime los pospotenciales tempranos inducidos en situaciones en que aumenta la amplitud de la INaL (isquemia, hipoxia, exposición a la toxina ATX-II o a radicales libres)47,50,52,53,56.
La ranolazina no produce arritmias en modelos experimentales en que los fármacos que prolongan el intervalo QTc (quinidina, ATX-II, cisaprida, moxifloxacino, ziprasidona) inducen la aparición de arritmias ventriculares; de hecho, en estas circunstancias, la ranolazina suprime los pospotenciales tempranos y las arritmias ventriculares inducidas por esos fármacos50,52-55,74-76. Más aún, en modelos animales con síndrome de SQTL3 que presentan un aumento de la INaL, la ranolazina reduce la INaL, acorta la DPA y el intervalo QTc, suprime los pos potenciales tempranos y las arritmias que éstos producen45,49,50,52,54,55,77.
La razón por la que en todos estos modelos la ranolazina no produce (e incluso suprime) arritmias cardiacas es que la posible prolongación de la DPA producida por el bloqueo de la IKr sería contrarrestada por el bloqueo de la INaL que el fármaco produce41,42,45,52-56,73,76. Ello explicaría por qué en ensayos clínicos controlados el intervalo QTc aumenta 1,9 y 4,9ms a la dosis de 500–750mg/12h60,61,73,78.
La ranolazina no modifica la INa, lo que explica por qué no modifica el complejo QRS ni la velocidad de conducción intracardiaca. Tampoco modifica la corriente lenta de entrada de Ca2+ a través de los canales L de las células musculares cardiacas o lisas vasculares, razón por la que el fármaco no modifica el intervalo PR del ECG, la frecuencia y la contractilidad cardiacas o las resistencias vasculares periféricas y la presión arterial.
Acciones antiarrítmicas. La ranolazina previene las arritmias ventriculares inducidas en un modelo canino de síndrome de QT largo54 y previene/termina las torsades de pointes en diversos modelos animales por fármacos que bloquean la corriente IKr-HERG12,45,50,55. En el estudio MERLIN-TIMI 36, que incluyó a 6.560 pacientes con síndrome coronario agudo sin elevación del segmento ST, la ranolazina disminuyó la incidencia de taquicardias ventriculares de más de 8 latidos (el 5,3 frente al 8,3%; p<0,001), taquicardias supraventriculares≥120lpm (el 44,7 frente al 55%; p<0,001) y pausas sinusales<3s (el 3,1 frente al 4,3%; p=0,01) y parece que redujo la incidencia de nuevos episodios de fibrilación auricular (el 1,7 frente al 2,4%; p=0,08)64. Los efectos antiarrítmicos ventriculares de la ranolazina persisteron en pacientes con disfunción ventricular (fracción de eyección<45%), una puntuación de riesgo TIMI de 5–7, insuficiencia cardiaca y prolongación basal del intervalo QTc.
Ranolazina en pacientes con síndrome de QT largo congénito. Los pacientes con síndrome de QT largo tipo 3 (SQTL3) presentan una mutación en el gen SCN5A que codifica la expresión de la subunidad alfa del canal de Na+ cardiaco, que no se inactiva o se reactiva de forma repetitiva e incrementa la amplitud de la INaL; ello explica la prolongación de la DPA ventricular y del intervalo QTc, así como la aparición de pospotenciales tempranos y torsades de pointes18. En cardiomiocitos que expresan una variante del SQTL3 (la deleción ¿KPQ1505-1507, situada en el lazo entre los dominios III y IV) la ranolazina bloquea el aumento de la INaL (IC50=15μmol) y acorta de la DPA ventricular49,77. Recientemente se ha demostrado en 5 pacientes con SQTL3 que presentaban la deleción ¿KPQ1505- 1507 que la infusión intravenosa de ranolazina (45mg/h durante 3h seguidos de 90mg/h durante 5h) acortaba el intervalo QTc (de 558±55 a 532±46ms; p<0,0001) sin prolongar los espacios PR y QRS y mejoraba la relajación ventricular, con un efecto lusitrópico positivo79. Así, la ranolazina acortó el tiempo de relajación isovolumétrica del ventrículo izquierdo (de 125±27 a 109±35ms) y aumentó la velocidad pico de la onda E de flujo mitral (de 57±8 a 71±9cm/s)79. Ese estudio es la primera evidencia de que la ranolazina inhibe la INa en el ser humano y confirma que el aumento patológico de la INaL está implicado en alteraciones eléctricas (prolongación del QTc, arritmias ventriculares) y contráctiles (aumento de la rigidez ventricular) en el miocardio humano.
Diabetes mellitus. Los pacientes diabéticos con angina crónica estable presentan un peor pronóstico80. En pacientes diabéticos con angina crónica estable81 o síndrome coronario agudo82, la efectividad antianginosa de la ranolazina era similar a la observada en no diabéticos, pero los pacientes tratados con el fármaco (750mg/12h) presentaron una reducción del 0,5% en la concentración de HbA1c. Además, la ranolazina aumentó el porcentaje de pacientes con HbA1c<7% respecto al grupo placebo (del 49 al 59%; p=0,004) y en pacientes no diabéticos redujo la incidencia de valores basales de glucosa>110mg/dl o los de HbA1c≥6% durante el estudio (el 31,8 frente al 41,2%; p=0,003)82.
El mecanismo de esta reducción de la HbA1c es desconocido, pero en modelos experimentales la ranolazina aumenta la liberación de insulina estimulada por glucosa por las células beta pancreáticas83 y mejora la homeostasis de la glucosa en ratas alimentadas con sucrosa, un modelo de resistencia a la insulina. Sin embargo, la ranolazina no modifica los valores plasmáticos basales de glucosa o de insulina y, a diferencia de otros insulinotropos, no produce episodios de hipoglucemia en pacientes anginosos82. Actualmente se desconoce si los efectos de la ranolazina en pacientes diabéticos son consecuencia del bloqueo de la INaL o de cambios en las concentraciones de ATP y/o Ca2+ en las células beta pancreáticas.
Efectos en el metabolismo cardiaco. El metabolismo cardiaco se realiza a expensas de la betaoxidación de los ácidos grasos, proceso que conlleva un mayor consumo (15%) de O2 por molécula de ATP sintetizada que la glucolisis84. Por ello, la betaoxidación de los ácidos grasos no es eficaz en situaciones en que el aporte coronario de O2 disminuye, como durante la isquemia. La ranolazina inhibe la betaoxidación de los ácidos grasos en el miocardio y desplaza el metabolismo energético cardiaco hacia la glucolisis, lo que supone una mayor eficiencia energética8,59,85. Sin embargo, este efecto aparece a concentraciones supraterapéuticas, por lo que tendría un mínimo papel en la acción antianginosa de la ranolazina8,86.
Otras acciones. La ranolazina es un débil bloqueador de los receptores β1/β2-adrenérgicos y muestra propiedades agonistas de los receptores 5-HT1A y 5-HT273,87. Sin embargo, las acciones simpaticolíticas no parecen ser importantes en los efectos antianginosos/antiisquémicos de la ranolazina, ya que ésta no modifica la frecuencia cardiaca o la presión arterial8,60,61.
Propiedades farmacocinéticas y dosificaciónLa formulación de liberación retardada de ranolazina presenta una biodisponibilidad oral del 75% y alcanza las concentraciones plasmáticas máximas (1,8-6μmol/l) al cabo de 2–5h y las estables al cabo de 3–4 días8,60,61,73,88. Se une a proteínas plasmáticas (65%), principalmente a la glucoproteína α1 ácida, presenta un volumen de distribución de 2,5l/kg y se biotransforma en un 70-85% en el hígado a través del citocromo P450 CYP3A4 (en menos de un 20% por el CYP2D6); se elimina un 73% por vía renal (el 5% sin biotransformar) y un 23% por las heces73,89,90. Su semivida de eliminación es de 7h8,13,73,88. Las propiedades farmacocinéticas de la ranolazina son independientes de la ingesta de alimentos y del sexo y no se alteran en pacientes con diabetes mellitus, hipertensión arterial, enfermedad pulmonar obstructiva crónica o insuficiencia cardiaca12,13,73,88. El área bajo la curva de las concentraciones plasmáticas de ranolazina aumenta 1,2 veces en pacientes con un aclaramiento de creatinina entre 30 y 50ml/min, 1,6-2 veces en pacientes con insuficiencia renal (aclaramiento de creatinina<30ml/min)12,89, 1,3 veces en pacientes con insuficiencia cardiaca (clase funcional NYHA III-IV) y 1,8 veces en pacientes con insuficiencia hepática moderada12,13.
La dosis inicial de ranolazina es de 375mg dos veces al día, que se incrementará al cabo de 2–4 semanas hasta 500mg y, posteriormente, hasta 750mg dos veces al día. Las tabletas del fármaco deben tragarse enteras (sin masticar o partir).
Reacciones adversasEl perfil de seguridad de la ranolazina ha sido analizado en diversos ensayos clínicos a corto60-62 y largo plazo63,64,78. Debemos señalar que en esos estudios la mayoría de los pacientes recibían dosis de ranolazina superiores (1.000 o 1.500mg dos veces al día) a la recomendada en la UE (750mg dos veces al día). A pesar de ello, la ranolazina fue bien tolerada, y las reacciones adversas más frecuentes fueron mareos, astenia, náuseas, dispepsia y estreñimiento. La incidencia de estas reacciones adversas es mayor en los pacientes de edad>65 años61,62,81. A dosis de 2g/día, se han observado 5 casos de síncope, posiblemente relacionados con el bloqueo de los receptores alfaadrenérgicos60,61. Sin embargo, no se ha observado ningún caso de síncope a las dosis recomendadas (375–750mg/12h). De hecho, en el estudio MERLIN-TIMI 36 las incidencias de síncope en los grupos activo y placebo fueron del 3 y el 2,1%, respectivamente63,64. Se han observado ligeros aumentos en los valores plasmáticos de creatinina (> 0,1mg/dl), que se han atribuido a la inhibición de su secreción tubular, y no a un efecto indeseable en la función glomerular. En los estudios CARISA61 y MERLIN-TIMI 3682, la incidencia de reacciones adversas era similar en los pacientes diabéticos y no diabéticos. En el estudio MERLIN-TIMI 36, las incidencias de mortalidad total o cardiovascular y de muerte súbita cardiaca eran similares en el grupo tratado con ranolazina y el de placebo.
Dado que la ranolazina bloquea la IKr puede prolongar la duración del potencial de acción cardiaco y el intervalo QT del ECG. En el estudio MARISA, la prolongación del intervalo QTc (utilizando la fórmula de Bazett) era de 6 y 7ms en los pacientes tratados con 500 y 1.000mg/12h60 y en el CARISA, 6,1 y 9,2ms respectivamente61. En el estudio ROLE, la prolongación media del QTc era de 2,4ms (p<0,001) y se pudo calcular que la prolongación del intervalo QTc producido por la ranolazina era de 2,4ms cada 1.000ng/ml del fármaco, lo que se traduce en una prolongación de 2–7ms a la dosis de 500–1.000mg/12h13. No se observó ningún caso de torsades de pointes en el estudio ROLE78, y en el estudio MERLIN-TIMI 36 se describieron dos casos, uno en el grupo placebo y otro en el grupo tratado con ranolazina64.
Interacciones medicamentosas y contraindicacionesLa ranolazina se biotransforma a través de CYP3A4 y CYP2D6 y, además, es un débil inhibidor de CYP3A4 y CYP2D 614,73,88. Los fármacos que inhiben moderada (diltiazem, fluconazol, eritromicina) o marcadamente (ketoconazol, itraconazol, voriconazol, inhibidores de la proteasa HIV, claritromicina, nefazodona) el CYP3A4 pueden aumentar las concentraciones plasmáticas de ranolazina8. Por el contrario, los inductores de CYP3A4 (rifampicina, fenitoína, hierba de San Juan) reducen la eficacia antianginosa de la ranolazina, por lo que se debe evitar combinarlos. El diltiazem inhibe la metabolización y reduce la eliminación de ranolazina, con lo que aumentan sus concentraciones plasmáticas 1,5-2,4 veces; la ranolazina duplica las concentraciones plasmáticas de simvastatina y aumenta 1,5 veces las de digoxina, por lo que puede ser necesario reducir la dosis de éstas88,90. La ranolazina es sustrato de la glucoproteína P y los fármacos que la inhiben (verapamilo, ciclosporina, quinidina) incrementan las concentraciones plasmáticas de ranolazina8. No se recomienda la administración de ranolazina a mujeres durante el embarazo y la lactancia. No es necesario reajustar la dosis de ranolazina cuando se combina con fármacos que son sustrato del CYP2D6, pero puede ser necesario reducir la dosis de algunos fármacos (antidepresivos tricíclicos, antipsicóticos) que son sustrato de esta isoenzima13,73.
La ranolazina está contraindicada en pacientes con prolongación del intervalo QTc, insuficiencia hepática moderada o insuficiencia renal grave (aclaramiento de creatinina<30ml/min) o en tratamiento con inhibidores potentes del CYP3A4, antiarrítmicos de los grupos IA y III (excepto amiodarona) o que prolonguen el intervalo QTc14,88.
CONCLUSIONESLa INaL tiene un importante papel en la acumulación de Na+ y Ca2+, que contribuye a la disfunción mecánica, eléctrica y metabólica en el miocardio isquémico. Por lo tanto, la INaL es una nueva diana terapéutica en el tratamiento de la angina crónica estable. A diferencia de estos fármacos, el efecto antianginoso de la ranolazina no está relacionado con una reducción de las MVO2 o un aumento del flujo sanguíneo coronario, sino con el bloqueo de la activación de la INaL que se activa de forma patológica en el miocardio isquémico. Ello se traduce en una reducción de las concentraciones intracelulares de Na+y Ca2+ durante la isquemia, lo que conlleva una reducción de la tensión parietal ventricular durante la diástole y de las MVO2 y un aumento de la perfusión coronaria. Sin embargo, la ranolazina no modifica la frecuencia o la contractilidad cardiacas, la presión arterial o la conducción auriculoventricular. La ranolazina es un nuevo abordaje farmacológico de los pacientes con angina crónica estable que son refractarios o no toleran el tratamiento convencional con nitratos, bloqueadores beta y antagonistas del calcio. La eficacia y seguridad de la ranolazina han sido confirmadas en ensayos clínicos controlados realizados en pacientes con angina crónica estable o con síndromes coronarios agudos sin elevación del segmento ST. En estos pacientes, la ranolazina, además, mejora la función ventricular y reduce la incidencia de arritmias cardiacas y las concentraciones de HbA1c en pacientes diabéticos. Estos nuevos hallazgos amplían el espectro de futuras aplicaciones de la ranolazina, algo que deberá ser confirmado en futuros ensayos clínicos controlados.
CONFLICTO DE INTERESESLos autores declaran no tener ningún conflicto de intereses.