
La trombosis tiene un papel crucial en la patogenia del infarto agudo de miocardio, los accidentes cerebrovasculares y la tromboembolia venosa y es el principal factor de su desenlace fatal. Estas enfermedades, cada una de las cuales tiene una incidencia anual de 1-3/1.000 individuos adultos, constituyen una de las primeras causas de mortalidad y morbilidad en la sociedad occidental. En consecuencia, a su diagnóstico, su tratamiento y su prevención se dedican grandes esfuerzos asistenciales y económicos.
La trombosis es un buen ejemplo de enfermedad compleja, donde la acción de múltiples genes y su interacción con factores ambientales determinarán en cada individuo el grado de susceptibilidad a padecer la enfermedad. Está bien establecido que variantes genéticas en los genes que codifican para factores o inhibidores de la coagulación son importantes factores de riesgo tromboembólico; sin embargo, en el 50% de los pacientes con trombofilia hereditaria no se identifica ninguna de estas alteraciones. Por consiguiente, el gran reto en la actualidad es la identificación de nuevos factores genéticos de riesgo trombótico.
Palabras clave
En 1965 se reconoció que la trombosis podía ser una enfermedad heredable, cuando Egeberg describió una familia con trombofilia asociada con una deficiencia en antitrombina (AT)1. Desde entonces, el concepto de trombofilia ha evolucionado considerablemente. Dado que las deficiencias en proteínas de la coagulación que incrementaban el riesgo de eventos trombóticos, como la AT, la proteína C (PC) y la proteína S (PS), y la disfibrinogenemia2 seguían un patrón de herencia autosómico dominante, se pensó que la trombofilia familiar la causaba un defecto génico dominante con una reducida penetrancia. Esta observación explicaría la variable expresividad clínica de la enfermedad. Sin embargo, esta idea de la trombofilia familiar como una enfermedad monogénica (con un modelo de herencia mendeliana simple) quedó en entredicho a través del estudio sistemático de la deficiencia de la PC3. Como resultado de estos estudios, se hipotetizó que la trombofilia familiar seguía un modelo poligénico de herencia, donde la cosegregación de uno o más factores genéticos adicionales incrementarían el riesgo trombótico3. Esta hipótesis se confirmó después de la identificación en los años noventa de las mutaciones del factor V Leiden (FVL)4 y G20210A en la región 3’ no traducible del gen de la protrombina5 como factores de riesgo trombótico adicional en las familias portadoras de deficiencias en PC, PS o AT6,7.
El siguiente paso que ha revolucionado el estudio de la trombofilia es la evolución que ha sufrido el concepto de enfermedad, que ya no es una variable discreta, dicotómica (individuos sanos o individuos enfermos), sino una variable de riesgo continua. En otras palabras, lo que está regulado genéticamente no es la enfermedad en sí misma, sino la mayor o menor predisposición a padecerla.
Por todo lo expuesto, actualmente se considera que la trombofilia hereditaria es una enfermedad multifactorial (interacción de factores genéticos y ambientales) y compleja (no sigue una herencia mendeliana simple), en la que la suma de múltiples genes y cada uno de ellos interaccionando con factores ambientales determinan en cada individuo el grado de susceptibilidad a la trombosis8,9.
MÉTODOS DE ESTUDIO DE LA TROMBOFILIASin embargo y pese a los grandes esfuerzos invertidos en la última década en el estudio de la enfermedad trombótica, nuestros conocimientos sobre la base molecular de esta afección son escasos, más si se tiene en cuenta que el 60% de la predisposición a la trombosis es atribuible a factores genéticos10. Para ilustrar esta situación basta recordar que los factores genéticos de riesgo trombótico conocidos sólo se identifican, en el mejor de los casos y dependiendo de la población estudiada, en el 50% de las familias con trombosis hereditaria. El gran reto en la actualidad es la identificación de los factores genéticos de riesgo trombótico en el 50% restante.
Los diseños experimentales utilizados en la identificación y la localización de genes implicados en el riesgo de trombosis incluyen análisis de ligamiento genético y los estudios de asociación.
Estudios de asociaciónLos estudios de asociación de casos y controles analizan la correlación entre fenotipo y genotipo. El fenotipo es generalmente la presencia (casos) o ausencia (controles) de la enfermedad en individuos no relacionados o en familias. El genotipo está determinado por algún tipo de polimorfismo genético, principalmente variaciones de un solo nucleótido, conocidas como single nucleotide polymorphism (SNP). Existe asociación cuando la distribución (frecuencias alélicas) de este marcador es diferente en los casos y los controles para un determinado nivel de significación estadística. El marcador genético es un polimorfismo dentro o cerca de un gen candidato, definido como un gen específico relacionado biológicamente con el proceso de la enfermedad. Este tipo de diseño fue el usado para identificar el FVL4 y la mutación G20210A en el gen de la protrombina5. Más recientemente, también ha permitido la identificación de las mutaciones A384S en el gen de la SERPINAC111, que codifica para la AT, y la R67X en el gen SERPINC10 que codifica el inhibidor de la proteína Z12 asociadas a un incremento del riesgo de eventos trombóticos.
Por otro lado, el análisis de ligamiento genético difiere de los estudios de asociación en que se basa en la transmisión de los padres a su descendencia (cosegregación) de un marcador genético y una variante genética funcional. La cosegregación de un marcador genético sólo se puede detectar observando cómo se heredan los cromosomas de una generación a otra, lo que implica necesariamente el reclutamiento de individuos emparentados en familias.
Los análisis de ligamiento genético han permitido identificar la mutación C46T en el gen del F12 como factor de riesgo trombótico13. Este resultado se ha replicado en diversos estudios de asociación de casos y controles. Por otro lado, aunque se había reportado la posible implicación del grupo sanguíneo ABO como factor de riesgo trombótico, la primera evidencia genética de esta implicación vino de un estudio de ligamiento genético14. Posteriormente, diversos estudios de asociación han confirmado este resultado y han determinado que es el A1 el alelo que aumenta el riesgo de eventos trombóticos15,16.
Estudios de ligamiento genéticoSin embargo, es en el estudio de los determinantes genéticos de fenotipos intermediarios con la enfermedad tromboembólica donde los estudios de ligamiento genético están generando los resultados más destacables (tabla 1)58-64. La base de estos métodos descansa en la genética de rasgos cuantitativos analizados en familias. Los efectos genéticos se cuantifican en términos de heredabilidad (h2), que es la proporción de la variancia en un fenotipo atribuible exclusivamente al efecto de los genes. En la tabla 267-72 se muestra la heredabilidad de varios componentes de la hemostasia. La estimación de la heredabilidad es un paso previo indispensable antes de intentar la localización de los genes, puesto que si el fenotipo no tiene heredabilidad o ésta es muy pequeña (p. ej., < 10%), no tiene sentido práctico la búsqueda de genes. La herramienta matemática básica en este tipo de investigaciones es el análisis de la variancia, que permite separar el efecto debido a los factores genéticos del efecto causado por los factores ambientales que influyen en un rasgo o fenotipo cuantitativo y en la enfermedad compleja en estudio. Esta aproximación metodológica ha permitido determinar que más de un 60% de la predisposición a la trombosis es atribuible a factores genéticos10.
Resultados de análisis de todo el genoma basados en ligamiento genético (genome-wide linkage studies) en el ámbito de la enfermedad tromboembólica
| Fenotipo | Cromosoma | Gen | Referencia |
| FII-trombosis | 11p11 | F2 | 58 |
| FXII-trombosis | 5q35 | F12 | 13 |
| FXII | 10p13 | ? | 13 |
| Proteína S libre | 1q32 | C4BP | 59 |
| APCR/FVIII-trombosis | 18p11 | ? | 60 |
| Factor von Willebrand | 9p34 | ABO | 14 |
| Proteína C | 16 | NQO1 | 61 |
| Fibrinógeno | 12 | TCF1 | 42 |
| Fibrinógeno | 14 | ? | 42 |
| FVII | 13 | F7 | 56 |
| TFPI | 2 | TFPI | 62 |
| TAFI | 13 | CPB2 | 63 |
| FVIII | 5/11 | ? | 52 |
| Trombosis | 10p12 | ? | 64 |
| Trombosis | 18p11.2 | ? | 64 |
| Trombosis | 11q23 | ? | 64 |
?: gen candidato desconocido.
Una vez demostrado que un fenotipo es heredable, el siguiente paso es localizar los sitios cromosómicos (locus) que contienen genes que influyen en la variabilidad de ese fenotipo, cada uno de estos loci se conoce como QTL (quantitative trait locus). Un QTL puede explicar sólo una pequeña proporción de la variabilidad observada en un fenotipo, mientras que el resto se deberá a otros QTL y a los factores ambientales17. La localización de los QTL se consigue con el análisis de ligamiento genético. Existen varias estrategias metodológicas para realizar análisis de ligamiento genético, pero una de las más robustas y poderosas, desde el punto de vista estadístico, está basada en el análisis de los componentes de la variancia (variance-components linkage analysis). La idea es que parientes que se asemejen más en un determinado fenotipo deben de compartir más marcadores genéticos alrededor del gen que está influyendo en ese fenotipo, mientras que otros parientes más alejados del fenotipo en estudio no serían portadores de los mismos alelos18.
Para saber si dos loci están ligados, existen diferentes pruebas estadísticas. El parámetro clásico es la escala de LOD o logaritmo de la odds ratio (OR) entre dos probabilidades alternativas (LOD = log10 [probabilidad de que ambos loci estén ligados / probabilidad de que no lo estén]).
Este concepto es importante porque las técnicas más avanzadas de búsqueda de nuevos genes están basadas en la utilización de marcadores genéticos anónimos altamente polimórficos que, en caso de presentar ligamiento con fenotipos complejos, permiten detectar la presencia cercana del verdadero gen funcional.
Estos estudios han permitido identificar regiones en el genoma que determinan la variabilidad de los niveles de componente de la coagulación o, directamente, que determinan el riesgo de que se produzcan eventos tromboembólicos (tabla 1).
Tanto los análisis de ligamiento genético como los estudios de asociación tienen sus ventajas y sus inconvenientes. Los primeros generalmente son buenos para localizar nuevos genes, mientras que los segundos lo son para analizar genes conocidos. Por lo tanto, se puede considerar que ambos métodos son complementarios, y la cuestión no es tanto qué diseño utilizar, sino cuándo y cómo debe aplicarse cada método. Este punto constituye uno de los debates más controvertidos que actualmente hay en el campo de la genética de las enfermedades complejas19.
BASES GENÉTICAS DE LA TROMBOSISComo hemos comentado, las anomalías aceptadas como factores genéticos de riesgo trombótico son las deficiencias de AT, PC y PS y las mutaciones FVL y G20210A PT20 (tabla 3). Sin embargo, en los últimos años se ha avanzado en el conocimiento de la base genética de otros factores de riesgo de eventos tromboembólicos.
Resistencia a la proteína C activadaUno de estos avances hace referencia al fenotipo «resistencia a la proteína C activada» (APCR) descrito por Dahlback et al21 (1993), que se caracteriza por una baja actividad anticoagulante del sistema de la PC. Aunque se ha identificado la mutación FVL como principal causa de esta alteración plasmática4, todavía un 10-20% de los casos de APCR no son portadores de la mutación FVL, lo cual indica la presencia de otras mutaciones que causan el mismo fenotipo. Entre ellas, FV Cambridge y FV Hong Kong son variantes genéticas que afectan al aminoácido Arg306, y que también alteran la activación del sistema de la PC22,23.
Mutación C46T en el gen del F12Mucho más relevante es el caso de la mutación C46T en el gen del F12. El factor XII (FXII) es esencial en el inicio de la cascada de la coagulación sanguínea. La concentración de esta proteína en plasma tiene una significativa correlación genética positiva (r = 0,351) con la enfermedad tromboembólica24. A partir de un análisis integral del genoma (genome-wide scan), se demostró la implicación del gen estructural F12 en la determinación de la variabilidad plasmática de factor XII (LOD score = 4,76; p = 1,5 × 10-6) y la susceptibilidad a los eventos trombóticos24. Estudios posteriores de asociación de casos y controles han confirmado que la mutación C46T del F12 es un factor de riesgo de trombosis venosa25,26 o arterial27-29. Concretamente, los portadores homocigotos del alelo T tienen 5 veces más riesgo de eventos tromboembólicos que los no portadores. Recientemente, en la misma dirección, se ha publicado un estudio30 que muestra la implicación de este polimorfismo como factor de riesgo trombótico durante el primer embarazo en un seguimiento de 32.463 mujeres previamente asintomáticas.
Grupo sanguíneo ABOPor otro lado, se conoce desde finales de los años sesenta que los portadores del grupo sanguíneo distinto de O presentan un riesgo entre 2 y 4 veces superior de padecer eventos trombóticos. Esta relación se ha establecido a través de la asociación observada entre el grupo ABO y los niveles de factor VIII31, por una parte, y los de factor von Willebrand (FvW) por otra32, ya que las concentraciones de estas proteínas están claramente relacionadas con riesgo cardiovascular33. Un paso más en el conocimiento del grupo sanguíneo como factor de riesgo cardiovascular ha sido la demostración de que es el alelo A1 el que implica más riesgo de trombosis34. Recientemente, estos resultados se han confirmado en un metaanálisis que concluye que los pacientes portadores de los genotipos distintos de O tienen un riesgo de eventos trombóticos mayor que el de los portadores de los genotipos del grupo sanguíneo O16. Además, parece que haya un efecto sinérgico entre los genotipos distintos de O y la mutación FVL, y se incrementa 23 veces el riesgo (intervalo de confianza [IC] del 95%, 9,1-59,3) de sufrir acontecimientos trombóticos en los portadores distintos de O y FVL que en los del sanguíneo O sin FVL15. Estos resultados avalan la implicación del grupo sanguíneo ABO en el riesgo tromboembólico y su papel sinérgico con otros factores genéticos implicados en esta enfermedad.
Mutación (R67X) en el gen SERPINA10Recientemente, en un estudio multicéntrico español19, se ha identificado una nueva mutación (R67X) en el gen SERPINA10 (que codifica para el inhibidor de la proteína Z) como un importante factor de riesgo tromboembólico. El inhibidor de la proteína Z es una proteína de la familia de la serpinas que se considera un nuevo miembro del sistema hemostático, debido a su actividad anticoagulante inhibiendo los factores X y XI activados. Los portadores de esta mutación (R67X) tiene un riesgo 3,3 veces superior de padecer un evento trombótico que los no portadores19, comparable al riesgo de los portadores de la mutación FVL o la G20210A en el gen del F2. Además, esta mutación presenta una fuerte asociación con historia familiar de trombosis (p < 0,001).
Mutación A384S en el gen SERPINC1En el mismo estudio multicéntrico español18, se ha identificado otra mutación, la A384S en el gen SERPINC1 (que codifica para AT) como un importante factor de riesgo tromboembólico. Los portadores de esta mutación tienen un riesgo de padecer un evento trombótico unas 10 veces superior que los no portadores. Fenotípicamente, la A384S causa una deficiencia en AT muy peculiar, ya que presenta niveles antigénicos normales y actividad anti-FXa normal, pero una actividad anti-IIa reducida en presencia de heparina35. Según estas características, el efecto de la mutación A384S en la AT no puede detectarse con los métodos plasmáticos utilizados habitualmente en los laboratorios clínicos. Además, es importante destacar la relevancia clínica de la identificación de los pacientes portadores de esta mutación para una correcta administración del tratamiento anticoagulante ya que, debido al defecto que esta mutación causa en la proteína AT, las heparinas no fraccionadas pueden ser ineficientes como tratamiento anticoagulante. Por lo tanto, la detección de esta alteración genética es imprescindible y supone un avance significativo en el diagnóstico de las deficiencias de AT.
Mutación V34L en el gen del F13Por último, existen pruebas sobre la implicación de la mutación V34L en el gen del F13 en la enfermedad tromboembólica. La estabilización de las moléculas de fibrina por el factor XIII activado (FXIIIa) es un proceso esencial para la formación del coágulo. Este proceso tiene un mecanismo de retroalimentación positiva donde la fibrina activa al FXIII. Esta activación es más rápida cuando el aminoácido 34 del FXIII es una Leu que cuando es una Val36. Como consecuencia, esta alteración cambia la conformación de la fibrina que polimeriza formando una malla más delgada en el coágulo, con poros más pequeños y alterando las características de permeabilidad del coágulo36. Varios estudios han descrito un efecto protector del alelo Leu34 respecto al riesgo de tromboembolia37,38, y su consistencia se ha confirmado en un metaanálisis39. Al no existir ninguna determinación plasmática que detecte el efecto funcional de la variante Leu34 en la formación del coágulo, la detección de esta alteración genética supone un avance significativo en el diagnóstico de la enfermedad tromboembólica.
Otros factores de riesgo trombóticoOtros factores de la coagulación o del sistema fibrinolítico pueden estar implicados en la predisposición a la trombosis. En este sentido se ha descrito que concentraciones de fibrinógeno elevadas se asocian con un incremento del riesgo trombótico40; sin embargo, sólo un 5-9% de la variabilidad de esas concentraciones se explica por polimorfismos localizados en el gen de la cadena beta, y sólo un 4,2% está determinado por polimorfismos en el gen de la cadena alfa41. Estos resultados implican que otros factores genéticos son la causa de la mayor parte de la variación cuantitativa de este fenotipo. Recientemente, se han localizado dos regiones del genoma relacionadas con la concentración plasmática de fibrinógeno (tabla 1), una en el cromosoma 12 y otra en el 1442.
Desde el punto de vista genético, son muchos los polimorfismos en diferentes genes candidatos (la mayoría de los que codifican las proteínas anteriormente citadas) que están siendo sometidos a múltiples estudios de asociación para determinar su implicación en la enfermedad tromboembólica. Destacaríamos diversos polimorfismos en el gen del receptor endotelial de la PC (EPCR)43-45, polimorfismos en los genes que codifican para receptores plaquetarios46,47 y polimorfismos en el gen que codifica el TAFI, que a través de la determinación de la concentración plasmática de TAFI podrían modular el riesgo de trombosis48. La verdadera participación de estos polimorfismos en la enfermedad tromboembólica, en el mejor de los casos, está por demostrar o es controvertida por la aparición de resultados contradictorios49,50.
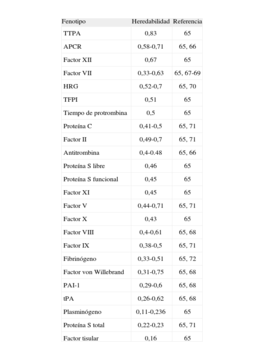
Aparte de los factores e inhibidores de la coagulación sanguínea anteriormente expuestos, otros factores de la hemostasia pueden estar implicados en la predisposición a la trombosis. Estos parámetros representan un buen ejemplo de fenotipo cuantitativo complejo, en el que las concentraciones en plasma son el resultado de la influencia combinada de factores genéticos y ambientales. Pero son los factores genéticos los que determinan una mayor proporción de estos factores; en otras palabras, existe una muy considerable base genética para la mayor parte de estos fenotipos (tabla 2).
Heredabilidad de los componentes de la hemostasia (parámetros de la coagulación y la fibrinolisis)
| Fenotipo | Heredabilidad | Referencia |
| TTPA | 0,83 | 65 |
| APCR | 0,58-0,71 | 65, 66 |
| Factor XII | 0,67 | 65 |
| Factor VII | 0,33-0,63 | 65, 67-69 |
| HRG | 0,52-0,7 | 65, 70 |
| TFPI | 0,51 | 65 |
| Tiempo de protrombina | 0,5 | 65 |
| Proteína C | 0,41-0,5 | 65, 71 |
| Factor II | 0,49-0,7 | 65, 71 |
| Antitrombina | 0,4-0.48 | 65, 66 |
| Proteína S libre | 0,46 | 65 |
| Proteína S funcional | 0,45 | 65 |
| Factor XI | 0,45 | 65 |
| Factor V | 0,44-0,71 | 65, 71 |
| Factor X | 0,43 | 65 |
| Factor VIII | 0,4-0,61 | 65, 68 |
| Factor IX | 0,38-0,5 | 65, 71 |
| Fibrinógeno | 0,33-0,51 | 65, 72 |
| Factor von Willebrand | 0,31-0,75 | 65, 68 |
| PAI-1 | 0,29-0,6 | 65, 68 |
| tPA | 0,26-0,62 | 65, 68 |
| Plasminógeno | 0,11-0,236 | 65 |
| Proteína S total | 0,22-0,23 | 65, 71 |
| Factor tisular | 0,16 | 65 |
APCR: resistencia a la proteína C activada; HRG: glucoproteína rica en histidina; PAI-1: inhibidor del activador del plasminógeno tipo 1; TFPI: inhibidor de la vía del factor tisular; t-PA: activador tisular del plasminógeno; TTPA: tiempo de tromboplastina parcial activado.
Entre estos fenotipos destacaríamos los niveles de FVIII. En estudios de asociación de casos y controles, se ha relacionado este fenotipo con el aumento de riesgo trombótico, con un riesgo relativo superior a 4 veces en los pacientes que alcanzan o superan las 150 U/dl de FVIII en plasma51. Actualmente, y como ya hemos explicado en el apartado anterior, el único determinante genético claramente implicado en la determinación de los valores plasmáticos de FVIII es el grupo sanguíneo ABO14,31. Pero ya disponemos de datos científicos que avalan la presencia de un gen (aún por identificar) en el cromosoma 552, que podría estar implicado en la determinación de los niveles de FVIII (tabla 1).
También se ha demostrado que tanto cifras por encima del percentil 90 de FXI como de FIX se asocian con incrementos del riesgo trombótico de 2,2 y unas 2-3 veces, respectivamente53,54 (tabla 3).
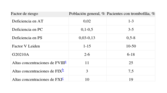
Prevalencia de alteraciones de la coagulación asociadas a riesgo tromboembólico
| Factor de riesgo | Población general, % | Pacientes con trombofilia, % |
| Deficiencia en AT | 0,02 | 1-3 |
| Deficiencia en PC | 0,1-0,5 | 3-5 |
| Deficiencia en PS | 0,03-0,13 | 0,5-8 |
| Factor V Leiden | 1-15 | 10-50 |
| G20210A | 2-6 | 6-18 |
| Altas concentraciones de FVIIIa | 11 | 25 |
| Altas concentraciones de FIXb | 3 | 7,5 |
| Altas concentraciones de FXIc | 10 | 19 |
AT: antitrombina; PC: proteína C; PS: proteína S.
Estudios de ligamiento genético en familias grandes han mostrado una fuerte correlación genética entre la susceptibilidad a los acontecimientos trombóticos y varios fenotipos de la hemostasia. Concretamente, los niveles de APCR, FVIII, FIX, FXI, FXII, FvW, activador tisular del plasminógeno (t-PA), generación de trombina, FVII, folato sérico y homocisteína están determinados por genes que a su vez incrementan el riesgo de eventos trombóticos10.
Dada la clara implicación de los niveles de los factores de la coagulación en el aumento de riesgo trombótico, la identificación de los factores genéticos que determinan estos niveles constituye una de las grandes líneas de investigación en el campo de la enfermedad tromboembólica55. En la actualidad, gracias a los estudios integrales del genoma, disponemos de información sobre regiones del genoma que contienen genes que determinan la variabilidad de diversos componentes de la coagulación (tabla 1). Incluso en algunos casos ya conocemos qué gen y qué variantes alélicas del gen son la causa de esta variabilidad y su posible implicación en la enfermedad tromboembólica13,56. En otros casos, aún nos queda un largo camino para identificar estos factores genéticos55.
CONCLUSIONESA través de esta revisión ha quedado bien establecido que la trombofilia es una enfermedad multifactorial compleja, en la que múltiples interacciones entre factores genéticos y ambientales contribuyen al desarrollo de la enfermedad. Sin embargo, pese a los grandes esfuerzos invertidos en la última década al estudio de la enfermedad trombótica, nuestros conocimientos sobre la base molecular de esta afección son escasos y sólo conocemos seis o siete factores genéticos que incrementan el riesgo de trombosis. Estos defectos genéticos sólo explican una parte pequeña de los casos de trombofilia hereditaria. Además, es improbable que estos defectos genéticos conocidos, con sus bajas frecuencias en la población, constituyan la influencia genética primaria del riesgo de que se desencadene un evento trombótico. El gran reto actual de los investigadores es la identificación de nuevos factores genéticos que contribuyan a la variación interindividual del riesgo trombótico.
La perspectiva futura es generar una lista de todos los factores genéticos que contribuyen a los eventos trombóticos. Este conocimiento ayudará a diseñar estrategias de tratamiento y prevención a medida del perfil genético del individuo57.
Actualmente, con la automatización de los métodos de genotipificación de marcadores genéticos y secuenciación de ADN, con la información proporcionada por el proyecto Genoma Humano y, principalmente, por el gran avance de la estadística genética y del poder de cálculo que proporciona la informática moderna, tenemos las herramientas necesarias y estamos en condiciones de abordar con éxito el reto que supone el estudio de la base genética que subyace a los rasgos complejos, como son la enfermedad tromboembólica y los fenotipos intermediarios que influyen en el riesgo de sufrir esta enfermedad. La identificación de estos nuevos factores genéticos y el estudio de su mecanismo fisiopatológico constituyen un paso fundamental en la comprensión de las bases moleculares de la trombosis. La investigación básica es imprescindible para el desarrollo de métodos diagnósticos, profilácticos y terapéuticos más eficaces. Además, este conocimiento conlleva ventajas asistenciales muy claras: mejorar la estrategia preventiva y terapéutica en los pacientes contra situaciones de riesgo y episodios trombóticos futuros e identificación de familiares afectos, la mayoría de ellos asintomáticos, que de otra forma no se beneficiarían de esta prevención.
Declaración de conflicto de interesesEl autor declara no tener conflicto de intereses.