
La muy esperada guía ESC 2023 sobre el tratamiento de las miocardiopatías es un hito fundamental en el tratamiento de estas enfermedades. Hasta ahora, los recursos disponibles se limitaban a varios documentos de consenso y una guía dedicada exclusivamente a la miocardiopatía hipertrófica1. Este documento se erige como la primera referencia exhaustiva que aborda las diversas miocardiopatías tanto a nivel global como con un enfoque específico2. Por la baja prevalencia de estas afecciones, la mayor parte de la información accesible procede de estudios observacionales y solo 5 recomendaciones están respaldadas por evidencia de nivel A.
NOVEDADESAl tratarse de la primera guía sobre este tipo de afecciones, muchas recomendaciones son novedosas. Entre los aspectos más destacados figuran los siguientes:
Clasificación de las miocardiopatíasSe introduce una nueva clasificación basada en las características morfológicas y los aspectos funcionales con el objetivo de estandarizar la nomenclatura y evitar la confusión que ha imperado en los últimos años. La guía distingue entre:
- -
Miocardiopatía hipertrófica (MCH), caracterizada por hipertrofia del ventrículo izquierdo en ausencia de causas secundarias.
- -
Miocardiopatía dilatada (MCD), caracterizada por dilatación y disfunción de los ventrículos sin causas identificables.
- -
Miocardiopatía no dilatada del ventrículo izquierdo (MCNDVI), una nueva entidad marcada por la existencia de fibrosis no isquémica (realce tardío de gadolinio [RTG]) o reemplazo del ventrículo izquierdo (VI) por tejido adiposo o disfunción sistólica sin dilatación. Este término abarca afecciones como la miocardiopatía hipocinética no dilatada, la miocardiopatía arritmogénica del ventrículo izquierdo (MCAVI) y la miocardiopatía no compactada (MCNC).
- -
Miocardiopatía arritmogénica del ventrículo derecho (MCAVD), con compromiso predominante del ventrículo derecho (VD), incluso con afectación biventricular. Se mantienen los criterios diagnósticos de 2010, mientras que no se incluyen los criterios de Padua por falta de validación externa3.
- -
Miocardiopatía restrictiva (MCR), disfunción diastólica con agrandamiento biauricular sin causa identificable y en ausencia de criterios para otra miocardiopatía.
Se centra en el concepto de que el fenotipo predominante en la presentación define la miocardiopatía y todas las genocopias entran dentro de esta clasificación morfofuncional. Se desaconseja el uso de MCNC (por su definición no morfofuncional) y MCAVI, haciendo hincapié en que la hipertrabeculación es más un rasgo fenotípico que una miocardiopatía en sí misma, ya que puede aparecer en población sana. Además, la guía reconoce que las miocardiopatías pueden manifestarse en contextos clínicos específicos que requieren una evaluación personalizada (p. ej., MCD alcohólica, periparto, asociada a la quimioterapia o miocarditis).
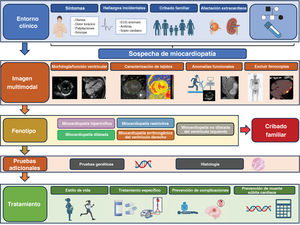
Mejora del diagnósticoSe introduce un planteamiento sistemático para establecer el tipo de miocardiopatía y alcanzar un diagnóstico etiológico (figura 1). La guía destaca la importancia de realizar un árbol genealógico de 3 generaciones, evaluar los signos/síntomas cardiacos y extracardiacos, realizar un electrocardiograma (ECG), colocar un Holter-ECG, hacer un análisis de sangre y llevar a cabo estudios de imagen multimodal, con tablas útiles basadas en otros documentos. Se recomienda la medición de la troponina y la fracción aminoterminal del propéptido natriurético cerebral (NT-proBNP), ya que pueden tener valor diagnóstico y pronóstico. Los pacientes pediátricos deben evaluarse con el mismo enfoque, reconociendo que los menores de 1 año presentan un perfil etiológico diferente.

La guía ha incorporado los avances en las técnicas de diagnóstico por imagen, propone realizar una resonancia magnética cardiaca (RMC) en el momento del diagnóstico en todos los casos, con una recomendación de nivel IB, y considera su repetición cada 2-5 años (IIaC). Además, se recomienda la RMC de aquellos miembros de la familia con genotipo positivo/fenotipo negativo para una detección temprana del fenotipo (IIaB).
Pruebas genéticas y asesoramientoSe destaca la importancia de las pruebas genéticas, ya que tienen valor diagnóstico, pronóstico y terapéutico en numerosas miocardiopatías. Los detalles específicos hacen referencia al documento de consenso de la European Heart Rhythm Association (EHRA) de 20224, con una tabla en que se relacionan todos los genes validados o candidatos de varias miocardiopatías. Se recomiendan las pruebas genéticas siempre que tengan impacto en la atención del paciente o ayuden a la evaluación familiar (clase IB). Se destaca el papel de la autopsia molecular en los diagnósticos post mortem.
Además, la guía estandariza el seguimiento de familiares, con recomendaciones hasta los 50 años de casos aparentemente esporádicos (estudio genético negativo y ningún otro familiar con miocardiopatía) y seguimiento a largo plazo de casos con más de un familiar afectado (con o sin mutación identificada, recomendación de clase IB y IIaC, respectivamente). Las variantes de significado incierto solo deben estudiarse si la segregación familiar permitiera la reclasificación (de novo o familia extensa). La edad a la cual se debe evaluar a los miembros de la familia pediátrica debe individualizarse.
TratamientoSe establecen principios generales aplicables a todas las miocardiopatías, como los tratamientos aprobados para la insuficiencia cardiaca (IC). Cabe destacar que la guía introduce la posibilidad de utilizar estos tratamientos en pacientes asintomáticos con fenotipo MCD/MCNDVI subclínico para prevenir la remodelación ventricular adversa (recomendación IIbC). Respecto a la fibrilación auricular (FA) o flutter, se recomienda la anticoagulación, independientemente de las puntuaciones de riesgo, para pacientes con MCH y amiloidosis (recomendación IB) o MCR (recomendación IIaC), haciendo gran hincapié en el control del ritmo.
El aspecto más novedoso y posiblemente controvertido corresponde a la prevención primaria de la muerte súbita cardiaca (MSC). El implante de desfibriladores automáticos (DAI) debe tratarse y acordarse con el paciente. Se recomiendan calculadoras de riesgo específico para MCH (IB), MCD, MCNDVI y MCAVD (IIaB).
Aspectos específicosMiocardiopatia hipertróficaLa guía principalmente se centra en aspectos novedosos, haciendo referencia a la guía de 2014 sobre aspectos específicos1. Entre las novedades notables se encuentran: a) la introducción del mavacamtén, un inhibidor de la miosina, como opción de tercera línea para el tratamiento de la MCH obstructiva sintomática, con una recomendación IIaA y b) la estratificación del riesgo en la prevención primaria de la MSC se basa en las puntuaciones de riesgo de MCH (tanto para adultos como para pediatría). La existencia de RTG >15% y la fracción de eyección del ventrículo izquierdo (FEVI) <50% se aceptan como factores modificadores en los grupos de riesgo bajo-moderado (se excluyen el aneurisma apical, la genética o la respuesta hipotensora al ejercicio en el proceso de toma de decisiones).
Miocardiopatía dilatada y miocardiopatía no dilatada del ventrículo izquierdoLa principal novedad radica en el valor de la genética y la caracterización tisular mediante RMC en la estratificación del riesgo de MSC, como se introdujo previamente en la guía de 2022 sobre arritmias ventriculares5. Los genes DSP y TMEM43 se añaden como genotipos de alto riesgo a los descritos con anterioridad (FLNC, RBM20, LMNA y PLN). El documento especifica las situaciones que requieren la consideración de un DAI con FEVI >35%. Se recomienda el uso de una calculadora de riesgo para LMNA, sin tener en cuenta los criterios clínicos clásicos. No obstante, no se ofrece ningún punto de corte específico. El documento también sugiere que se puede valorar el implante de un DAI en aquellos casos en que el estudio genético sea negativo cuando hay taquicardia ventricular no sostenida (TVNS), antecedentes familiares de MSC o RTG extenso.
Miocardiopatía arritmogénica del ventrículo derechoLa flecainida es una alternativa a la amiodarona en estos pacientes (IIaC). En la prevención primaria de la MSC, se recomienda el uso de factores de riesgo validados (TVNS, FEVI <45%, fracción de eyección del VD <40%), así como la calculadora de riesgo de MCAVD actualizada en 2019 para ayudar en el proceso de toma de decisiones. El documento reconoce que funciona mejor en pacientes con PKP2, pero no ofrece ningún punto de corte específico. El estudio electrofisiológico (inducción de TV) está reservado para pacientes sintomáticos.
Miocardiopatía restrictivaSe hace hincapié en la importancia de realizar un estudio etiológico preciso para administrar un tratamiento dirigido, en el cual la biopsia endomiocárdica es una herramienta fundamental. No hay recomendaciones sobre el implante de un DAI como prevención primaria de la MSC.
Recomendaciones generalesSe recomienda a los pacientes con miocardiopatía que realicen ejercicio con regularidad con una intensidad baja o moderada, con prescripción individualizada en función de la evaluación de riesgos (clase IC). En la mayoría de los casos, se permite a las personas participar en deportes de cualquier intensidad cuando la estratificación del riesgo de MSC sea baja, con un enfoque individualizado. Se mantiene la recomendación de evitar la práctica deportiva en MCAVD, incluso a los portadores sanos y a individuos con las variantes LMNA o TMEM43.
La guía recomienda evaluar el riesgo y ofrecer asesoramiento previo al embarazo mediante la clasificación de riesgo materno de la Organización Mundial de la Salud. Esto incluye información sobre los métodos anticonceptivos y el riesgo hereditario. Se recomienda el parto vaginal en la mayoría de los casos, si no existen indicaciones obstétricas de cesárea, insuficiencia cardiaca grave (FEVI <30% o clase III-IV de la New York Heart Association [NYHA]) u obstrucción grave del tracto de salida del VI. La seguridad de los medicamentos debe revisarse antes del embarazo, con ajustes basados en la tolerabilidad. Se debe considerar la posibilidad de continuar con los bloqueadores beta durante el embarazo, con una estrecha vigilancia del crecimiento fetal y de los recién nacidos si los beneficios superan los riesgos.
CONSECUENCIAS DE LA APLICACIÓN EN ESPAÑAImplicaciones diagnósticasSegún las nuevas recomendaciones, se espera que la solicitud de estudios de imagen cardiaca aumente considerablemente, sobre todo de RMC. Si bien esta técnica cada vez está más extendida, este aumento puede provocar retrasos excesivos en la evaluación inicial de los casos índice y de los familiares genéticamente positivos. Además, la guía recomienda repetir la RMC durante el seguimiento cada 2-5 años para controlar la progresión, la respuesta al tratamiento y la reestratificación del riesgo. La cuantificación de la extensión del RTG, incorporada como factor adicional en casos con riesgo de MSC bajo o intermedio según la puntuación propuesta, implica la necesidad de interpretar estos estudios con software específico, más allá de la presencia/ausencia de realce y su descripción, lo que los convierte en menos accesibles a nivel general.
En cuanto a las pruebas genéticas, los avances tecnológicos han reducido los costes y los plazos. El respaldo de la guía al papel de la genética en el diagnóstico y la estratificación pronóstica de las miocardiopatías aumentará la demanda, en especial en MCD y MCNDVI, con la propuesta de estratificación del riesgo de MSC con FEVI >35% basada en el genotipo (recomendación de clase IIaB). Dado que los recursos pueden ser limitados a veces, será útil estimar la probabilidad previa a la prueba para decidir a quién se debe dar prioridad. Por tanto, recursos como la puntuación de Madrid6 desempeñarán un papel cada vez más destacado.
Al mismo tiempo, un aumento de las pruebas genéticas también plantea el reto de interpretar las variantes que se encuentren, así como tratar correctamente la información, tanto de pacientes como de sus familiares, por parte de profesionales con conocimientos y experiencia en genética.
Implicaciones para el tratamiento médicoLa introducción de mavacamtén como alternativa a la disopiramida en pacientes con MCH obstructiva sintomática abre la puerta a su uso generalizado en nuestro entorno. Esta nueva alternativa terapéutica, aprobada por la Agencia Europea del Medicamento y la Agencia Española de Medicamentos y Productos Sanitarios, inicialmente estará disponible en España gracias a un programa de acceso anticipado como fármaco extranjero. Su aplicación en la práctica clínica requerirá el genotipado del citocromo CYP2C19 para definir la dosis adecuada7, un estrecho seguimiento con estudios ecocardiográficos para la titulación, con especial atención a la FEVI y la evaluación de la respuesta al tratamiento. Por tanto, la introducción de mavacamtén planteará un reto en la práctica sanitaria en España, sobre todo cuando sea accesible y reciba apoyo financiero.
Unidades especializadasEl tratamiento de las miocardiopatías requiere herramientas especializadas, amplia experiencia y un enfoque clínico-básico multidisciplinario. Aunque no existe ningún plan de estudios centralizado para cardiólogos especializados en miocardiopatías, la guía incorpora las habilidades y los requisitos recomendados para una unidad de miocardiopatía. En España, las acreditaciones SEC-EXCELENTE y Centros, Servicios y Unidades de Referencia (CSUR) garantizan que las personas con este cuadro clínico reciban el apoyo necesario y las indicaciones precisas por parte de profesionales expertos en la materia.
LAGUNAS EN EL CONOCIMIENTOEsta sección enumera una gran cantidad de áreas de conocimiento donde imperan más sombras que luces. Muchos de los temas destacados tienen su origen en la escasez de información exacta sobre miocardiopatías familiares, dada su condición de enfermedades raras (especialmente en niños), lo que complica la obtención de muestras de gran tamaño y la realización de ensayos clínicos.
Cabe destacar la incorporación del término novedoso MCNDVI, según el fenotipo, acompañado del consejo explícito de eliminar otros términos de entidades incluidas dentro de este (MCAVI y MCNC), que se basan en la histopatología. Este nuevo concepto, heterogéneo genética y mecanísticamente, aumenta la complejidad de las recomendaciones terapéuticas y la estratificación de las arritmias. Incluso el hecho de estimar su prevalencia es un desafío porque su diagnóstico requiere RMC. El nuevo sistema alinea a los cardiólogos con experiencia en miocardiopatías familiares con los expertos en imagen cardiaca, pero los aleja de los anatomopatólogos clínicos y forenses, que continuarán diagnosticando MCAVI en pacientes con diferentes diagnósticos clínicos antes de la muerte (MCNDVI).
Cabe destacar que la guía carece de una sección sobre miocarditis, teniendo en cuenta que no se ha publicado ningún documento de consenso desde 20138. A los autores del presente comentario les hubiera gustado una sección específica sobre este tema que ofreciera recomendaciones actualizadas para el diagnóstico y el tratamiento.
Los avances en las ciencias de la -ómica (proteómica, metabolómica y transcriptómica) ya están surgiendo en las miocardiopatías. De hecho, han esbozado nuevas subclasificaciones de MCD (grupos fibróticos, no fibróticos leves e insuficiencia biventricular), que podrían ser útiles para el diagnóstico y el pronóstico de las arritmias9. Estudios similares en pacientes con MCH han abierto nuevos horizontes para identificar nuevas dianas terapéuticas10.
Los avances importantes en las pruebas habituales (ECG, ecocardiografía y RMC) facilitarán diagnósticos más precoces y diagnósticos diferenciales con genocopias. Todavía hay lagunas, como el impacto futuro de la RMC en la detección, cómo estandarizar la cuantificación del RTG (idealmente automatizada) y cómo superar los obstáculos planteados por los portadores de dispositivos cardiacos, más allá de la mejora derivada de los cambios en la colocación del brazo11. Es probable que muchas soluciones provengan de la inteligencia artificial, que ya permite análisis precisos y masivos de ECG12.
La penetrancia incompleta de las miocardiopatías genéticas es actualmente materia de investigación y puede estar influida por mutaciones somáticas, mutaciones en regiones intrónicas de genes conocidos, en genes nuevos y aún desconocidos, ADN mitocondrial, el efecto aditivo en la puntuación conocida como «puntuación de riesgo poligénico» o cambios epigenéticos. La comprensión de estos fenómenos permitirá la personalización de la predicción de la evolución natural y la estratificación del riesgo. Probablemente, debido a la accesibilidad de los genomas completos en la población general, esta guía admite que se desconoce el impacto global de estos estudios en individuos sin indicación clínica.
En cuanto al tratamiento de las arritmias, se menciona la recomendación de flecainida para el control de FA en MCNDVI y para arritmias auriculares y ventriculares en MCAVD. Sin embargo, no se ofrecen detalles para tratar de superar la reticencia a utilizar flecainida ante cicatriz miocárdica, extrapolada de estudios antiguos sobre miocardiopatía isquémica. Se espera con impaciencia resultados en estimulación fisiológica13, ablaciones con sistemas de navegación que incorporan la caracterización de tejidos mediante imagen y la neuromodulación de arritmias malignas refractarias14. La prevención primaria de MSC aún necesita evidencia para mejorar la estimación del riesgo después de la reducción septal, en genotipos específicos y en MCD con FEVI recuperada con tratamiento.
La falta de evidencia también afecta a las recomendaciones deportivas y reproductivas. En general, las pautas deportivas son imprecisas y no definen limitaciones, excepto en MCAVD. Faltan detalles sobre el tipo y nivel de deporte permitidos. La definición de «individuos de bajo riesgo» y las recomendaciones para individuos con genotipo positivo/fenotipo negativo difieren de las de documentos anteriores de la ESC (que incluyen en los genotipos de alto riesgo FLNCtv y RBM20, además de LMNA y TMEM43). Respecto al embarazo, la falta de resultados de los registros contemporáneos continúa siendo una limitación importante para mejorar la estratificación del riesgo materno y fetal en la práctica diaria, seleccionar el fármaco antiarrítmico más adecuado y planificar el seguimiento. La única información sobre medicamentos y embarazo se muestra en la guía ESC 2018 sobre el embarazo e incluye solo evidencia preclínica y en modelos animales.
En cuanto al mavacamtén, se espera la confirmación en seres humanos de su efecto protector frente al desarrollo de fibrosis e hipertrofia, ya observado en ratones15, lo que podría ayudar a extender la indicación a individuos con genotipo positivo/fenotipo negativo. En la amiloidosis cardiaca por transtirretina existe una falta reconocida de evidencia que respalde el tratamiento con tafamidis en pacientes con afectación cardiaca y clase funcional III.
CONCLUSIONESEn resumen, la guía ESC 2023 sobre el tratamiento de las miocardiopatías es un hito importante en el tratamiento de estas afecciones. Estos comentarios tienen como objetivo resumir las novedades, resaltar las dificultades que conlleva su aplicación en el entorno clínico español e identificar las lagunas restantes en la evidencia. Con todo, se anima a los lectores a analizar el texto completo de la guía para profundizar en detalles específicos y mejorar su práctica diaria.
FINANCIACIÓNNinguna.
DECLARACIÓN SOBRE EL USO DE LA INTELIGENCIA ARTIFICIALDurante la preparación de la versión en inglés de este trabajo, los autores utilizaron ChatGPT para mejorar el lenguaje y la legibilidad. Después de utilizar esta herramienta, los autores revisaron y editaron el contenido en caso necesario, y asumen toda la responsabilidad por el contenido de la publicación.
CONFLICTO DE INTERESESLos documentos de declaración de conflicto de intereses de todos los autores aparecen en el material adicional.
Grupo de Trabajo de la SEC para la guía ESC 2023 sobre el tratamiento de las miocardiopatías: Eduardo Villacorta-Argüelles (coordinador), Laura Dos-Subirà (coordinadora), Francisco José Bermúdez-Jiménez, Esther González-López, José María Larrañaga-Moreira, Javier Limeres-Freire, Lidia López-García, Carmen Muñoz-Esparza, Esther Zorio-Grima.
Comité de Guías de la SEC: José Luis Ferreiro (presidente), Pablo Avanzas (secretario), Rut Andrea, Araceli Boraita, David Calvo, Raquel Campuzano, Victoria Delgado, Laura Dos Subirá, Juan José Gómez Doblas, María Antonia Martínez Momblan, Pilar Mazón, Domingo Pascual Figal, Juan Sanchis, José María de la Torre Hernández, David Vivas.
Los nombres de todos los autores del artículo figuran por orden alfabético en el anexo A.
Autor para correspondencia.
Correos electrónicos:evillacorta@secardiologia.es (E. Villacorta Argüelles); laura.dos@vallhebron.cat (L. Dos Subirà).