
The much-expected 2023 ESC Guidelines for the management of cardiomyopathies represent a pivotal milestone in the management of these diseases. Until now, the available resources were limited to various consensus documents and a guideline exclusively dedicated to hypertrophic cardiomyopathy.1 This document stands as the first comprehensive reference addressing the various cardiomyopathies both globally and with specific focus.2 Given the low prevalence of these conditions, most of the accessible information is derived from observational studies, with only 5 recommendations supported by level A evidence.
NOVELTIESBeing the first guidelines for these types of conditions, many recommendations are novel. Key highlights include the following:
Classification of cardiomyopathiesA new classification centered on morphological characteristics and functional aspects is introduced with the aim of standardizing the nomenclature and prevent the confusion that has emerged in recent years. The guidelines distinguish between:
- -
Hypertrophic cardiomyopathy (HCM), characterized by left ventricular hypertrophy in the absence of secondary causes.
- -
Dilated cardiomyopathy (DCM), characterized by ventricular dilation and dysfunction without identifiable causes.
- -
Nondilated left ventricular cardiomyopathy (NDLVC), a new entity marked by the presence of nonischemic fibrosis (late gadolinium enhancement, [LGE]) or fatty replacement in the left ventricle (LV) and/or systolic dysfunction without dilatation. This term encompasses conditions such as nondilated hypokinetic cardiomyopathy, left ventricular arrhythmogenic cardiomyopathy (LVAC), and noncompaction cardiomyopathy (NCC).
- -
Arrhythmogenic right ventricular cardiomyopathy (ARVC), with predominant compromise of the right ventricle (RV), even in the presence of biventricular involvement. The diagnostic criteria from 2010 are maintained, whereas the Padua criteria are not included due to the lack of external validation.3
- -
Restrictive cardiomyopathy (RCM), diastolic dysfunction with biatrial enlargement without an identifiable cause and in the absence of criteria for another cardiomyopathy.
Focus is placed on the concept that the predominant phenotype in presentation defines the cardiomyopathy, and all genocopies fall within this morphofunctional classification. The use of NCC (due to its nonmorphofunctional definition) and LVAC is discouraged, with an emphasis that hypertrabeculation is more of a phenotypic trait than a cardiomyopathy per se, as it can appear in a healthy population. Furthermore, the guidelines acknowledge that cardiomyopathies may manifest in specific clinical contexts that require tailored evaluation (eg, alcoholic DCM, peripartum, chemotherapy-associated, or myocarditis).
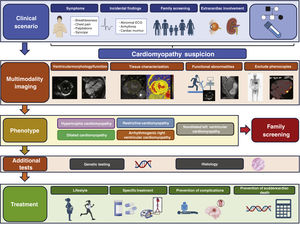
Enhanced diagnosisA systematic approach is introduced to establish the type of cardiomyopathy and reach an etiological diagnosis (figure 1). The guidelines highlight the importance of constructing a 3-generation family tree, assessing cardiac and extracardiac signs/symptoms, electrocardiogram (ECG), Holter-ECG, blood tests, and multimodal imaging studies, with useful tables based on other documents. The measurement of troponin and NT-proBNP is recommended as they may have diagnostic and prognostic value. Pediatric patients should be evaluated using the same approach, with recognition that those younger than 1 year exhibit a different etiological profile.

The guidelines have incorporated advances in imaging techniques, advocating for cardiac magnetic resonance (CMR) at diagnosis for all cases with a level IB recommendation and considering its repetition every 2 to 5 years (IIaC). Additionally, CMR is recommended for genotype-positive/phenotype-negative family members for an early detection of the phenotype (IIaB).
Genetic testing and counselingThe importance of genetic testing is stressed, as it holds diagnostic, prognostic, and therapeutic value in numerous cardiomyopathies. Specific details refer to the EHRA consensus document from 2022,4 with a table listing all validated or candidate genes for various cardiomyopathies. Genetic testing is recommended whenever it impacts patient care or helps in familial evaluation (class IB). The role of molecular autopsy in post-mortem diagnoses is underscored.
Additionally, the guidelines standardize the follow-up of family members, with recommendations up to the age of 50 years for apparently sporadic cases (negative genetic study and no other family members with cardiomyopathy) and long-term follow-up for cases with more than one affected family member (with or without identified mutation, recommendation class IB and IIaC, respectively). Variants of uncertain significance should only be studied if family segregation would allow for reclassification (de novo or extended family). The age at which to assess pediatric family members should be individualized.
TreatmentGeneral principles applicable to all cardiomyopathies are established, including approved treatments for heart failure (HF). Of note, the guidelines introduce the possibility of using these treatments in asymptomatic patients with subclinical DCM/NDLVC phenotype to prevent adverse ventricular remodeling (recommendation IIbC). Regarding atrial fibrillation (AF) or flutter, anticoagulation is recommended regardless of risk scores for patients with HCM and amyloidosis (recommendation IB) or RC (recommendation IIaC), with a strong emphasis on rhythm control.
The most novel and potentially controversial aspect pertains to primary prevention of sudden cardiac death (SCD). The implantation of automatic defibrillators (ICD) should be discussed and agreed upon with the patient. Specific risk calculators are recommended for HCM (IB), DCM, NDLVC, and ARVC (IIaB).
Specific aspectsHypertrophic cardiomyopathyThe guidelines primarily focus on new developments, referring to the 2014 guidelines for specific aspects.1 Notable novelties include: a) the introduction of the myosin inhibitor mavacamten as a third-line option for the management of symptomatic obstructive HCM, with a IIaA recommendation; and b) risk stratification for primary prevention of SCD relies on HCM-risk scores (both for adults and pediatrics). The presence of LGE >15% and left ventricular ejection fraction (LVEF) <50% are accepted as modifying factors in the low-moderate risk groups (excluding apical aneurysm, genetics, or hypotensive response on exercise in the decision-making process).
Dilated cardiomyopathy and nondilated left ventricular cardiomyopathyThe main novelty lies in the value of genetics and tissue characterization by CMR in risk stratification of SCD, as previously introduced in the 2022 guidelines for ventricular arrhythmias.5 The DSP and TMEM43 genes are added as high-risk genotypes to those previously described (FLNC, RBM20, LMNA, PLN). The document specifies the situations requiring consideration of an ICD with LVEF >35%. The use of a risk calculator for LMNA is recommended, disregarding classic clinical criteria. However, no specific cutoff point is provided. The document also suggests that ICD implantation may be considered in cases with a negative genetic study when nonsustained ventricular tachycardia (NSVT), family history of SCD, or extensive LGE are present.
Arrhythmogenic right ventricular cardiomyopathyFlecainide is an alternative to amiodarone in these patients (IIaC). In primary prevention of SCD, the use of validated risk factors (NSVT, LVEF <45%, RV ejection fraction <40%), as well as the 2019 updated ARCV-Risk calculator is recommended to aid the decision-making process. The document acknowledges that it works better in PKP2 patients but does not provide a specific cutoff point. Electrophysiological study (induction of VT) is reserved for symptomatic patients.
Restrictive cardiomyopathyThe importance of conducting a precise etiological study to provide targeted treatment is emphasized, with endomyocardial biopsy being a fundamental tool. There are no recommendations for ICD implantation as primary prevention of SCD.
General recommendationsPatients with cardiomyopathy are recommended to engage in regular exercise at low to moderate intensity levels, with individualized prescription based on risk assessment (class IC). In most cases, individuals are allowed to participate in sports of any intensity when the SCD risk stratification is low, with an individualized approach. The recommendation to avoid sports in ARVC, including healthy carriers and in individuals with LMNA or TMEM43 variants, is retained.
The guidelines recommend assessing risk and providing prepregnancy counseling using the World Health Organization maternal risk classification. This includes information on contraceptive methods and hereditary risk. Vaginal delivery is recommended in most cases, unless there are obstetric indications for cesarean section, severe heart failure (LVEF <30% or NYHA class III-IV), or severe LV outflow tract obstruction. The safety of medications should be reviewed before pregnancy, with adjustments made based on tolerability. Consideration should be given to continuing beta-blockers during pregnancy, with close monitoring of fetal growth and newborns if the benefits outweigh the risks.
CONSEQUENCES OF THE IMPLEMENTATION IN SPAINDiagnostic implicationsFollowing the new recommendations, the demand for cardiac imaging studies is expected to increase substantially, especially for CMR. While this technique is becoming more widely available, this increase may cause excessive delays in the initial evaluation of index cases and genetically positive family members. Furthermore, the guidelines recommend repeating CMR during follow-up every 2 to 5 years to monitor progression, treatment response, and risk restratification. The quantification of LGE extension, incorporated as an additional factor in cases with low or intermediate SCD risk according to the proposed score, implies the need to interpret these studies with specific software, beyond the presence/absence of enhancement and its description, making it less accessible in general.
Regarding genetic testing, technological advances have reduced costs and timeframes. The guidelines’ endorsement of the role of genetics in the diagnosis and prognostic stratification of cardiomyopathies will increase demand, particularly for DCM and NDLVC, with the proposal for SCD risk stratification with LVEF >35% based on genotype (class IIaB recommendation). As resources can be limited at times, it will be helpful to estimate the pre-test probability to decide whom to prioritize. Therefore, resources like the Madrid score6 will play an increasingly prominent role.
At the same time, an increase in genetic testing also poses the challenge of interpreting the variants found, as well as managing the information correctly, both in patients and their family members, by professionals with knowledge and experience of genetics.
Implications for medical treatmentThe introduction of mavacamten as an alternative to disopyramide in symptomatic obstructive HCM patients opens the door to its widespread use in our setting. This new therapeutic alternative, approved by the European Medicines Agency and the Agencia Española de Medicamentos y Productos Sanitarios, will initially be available in Spain through an early access program as a foreign drug. Its implementation in clinical practice will require genotyping of the cytochrome CYP2C19 to define the appropriate dose,7 close monitoring with echocardiographic studies for titration, with special attention to LVEF, and evaluation of treatment response. Therefore, the introduction of mavacamten will pose a challenge in health care practice in Spain, especially when it becomes financially supported and accessible.
Specialized unitsThe management of cardiomyopathies requires specialized tools, extensive experience, and a multidisciplinary basic-clinical approach. Although there is no centralized curriculum for cardiologists dedicated to cardiomyopathies, the guidelines incorporate recommended skills and requirements for a cardiomyopathy unit. In Spain, the SEC-EXCELENTE and Centros, Servicios y Unidades de Referencia (CSUR) certifications ensure that individuals with this condition receive the necessary support and precise guidance from professionals who are experts in the field.
KNOWLEDGE GAPSThis section lists a plethora of knowledge areas where shadows continue to prevail over light. Many of the highlighted items originate from the scarcity of precise information on familial cardiomyopathies, given their status as rare diseases (especially in children), which complicates obtaining large sample sizes and conducting clinical trials.
It is worth noting the incorporation of the novel term NDLVC, based on phenotype, accompanied by the explicit advice to eliminate other terms of entities included within it (LVAC and NCC), which are based on histopathology. This new concept, genetically and mechanistically heterogeneous, increases the complexity of therapeutic recommendations and arrhythmic stratification. Even estimating its prevalence is challenging because its diagnosis requires CMR. The new paradigm aligns cardiologists with expertise in familial cardiomyopathies with cardiac imaging experts but separates them from clinical and forensic pathologists, who will continue to diagnose LVAC in patients with different clinical diagnoses before death (NDLVC).
It should be emphasized that the guidelines lack a section on myocarditis, considering that no consensus document has been published since 2013.8 The authors of the present comment would have liked a specific section on this topic providing updated recommendations for diagnosis and treatment.
Advances in -omics sciences (proteomics, metabolomics, and transcriptomics) is already emerging in cardiomyopathies. Indeed, they have outlined new subclassifications of DCM (fibrotic groups, mild nonfibrotic, and biventricular failure), which could be useful for diagnosis and arrhythmic prognosis.9 Similar studies in HCM patients have opened up new horizons for identifying new therapeutic targets.10
Significant advances in routine tests (ECG, echocardiography, and CMR) will facilitate earlier diagnoses and differential diagnoses with genocopies. There are still gaps, such as the future impact of CMR on screening, how to standardize LGE quantification (ideally automated), and how to overcome the obstacles posed by cardiac device carriers, beyond the improvement resulting from changes in arm positioning.11 Many solutions will likely come from artificial intelligence, which already allows precise and massive ECG analyses.12
Incomplete penetrance of genetic cardiomyopathies is currently a matter of research and may be influenced by somatic mutations, mutations in intronic regions of known genes, in new, still unknown genes, mitochondrial DNA, the additive effect in the score known as the “polygenic risk score”, or epigenetic changes. Understanding these phenomena will allow for the customization of natural history prediction and risk stratification. Probably, due to the accessibility of complete genomes in the general population, these guidelines recognize that the global impact of these studies on individuals without clinical indication is unknown.
Regarding arrhythmic management, the recommendation of flecainide for AF control in NDLVC and for atrial and ventricular arrhythmias in ARVC is mentioned. However, no details are given to help to overcome the reluctance to use flecainide in the presence of myocardial scar, extrapolated from old studies on ischemic cardiomyopathy. Results are eagerly awaited in physiological stimulation,13 ablations with navigation systems that incorporate tissue characterization through imaging, and neuromodulation for refractory malignant arrhythmias.14 Primary prevention of SCD still needs evidence to improve risk estimation after septal reduction, in specific genotypes, and in DCM with recovered LVEF with treatment.
The lack of evidence also affects sports and reproductive recommendations. Overall, the sports guidelines are vague and do not define limitations, except in ARVC. Details about the type and level of sports allowed are lacking. The definition of “low-risk subjects” and recommendations for genotype-positive/phenotype-negative individuals differ from previous ESC documents (others include high-risk genotypes FLNCtv and RBM20, in addition to LMNA and TMEM43). Regarding pregnancy, the lack of results from contemporary registries remains a major shortcoming to improve maternal and fetal risk stratification in daily practice, select the most suitable antiarrhythmic drug, and plan the follow-up. The only information on drugs and pregnancy is shown in the 2018 ESC guidelines for pregnancy and includes preclinical and animal model evidence.
Regarding mavacamten, confirmation in humans of its protective effect on the development of fibrosis and hypertrophy, already observed in mice,15 is awaited, which could help to extend the indication to genotype-positive/phenotype-negative individuals. In cardiac transthyretin amyloidosis, there is a recognized lack of evidence supporting tafamidis treatment in patients with cardiac involvement and functional class III.
CONCLUSIONSIn summary, the 2023 ESC guidelines for the management of cardiomyopathies represent a significant milestone in the management of these conditions. These comments aim to summarize the novelties, highlight the challenges of their implementation in the Spanish clinical setting, and identify the remaining gaps in evidence. However, we encourage readers to analyze the full text of the guidelines to delve deeper into specific details and enhance their daily practice.
FUNDINGNone.
STATEMENT ON THE USE OF ARTIFICIAL INTELLIGENCEDuring the preparation of this work, the authors used ChatGPT to improve language and readability. After using this tool, the authors reviewed and edited the content as needed and take full responsibility for the content of the publication.
CONFLICTS OF INTERESTThe conflict-of-interest declaration documents of all authors can be seen in the supplementary data.
SEC Guidelines Committee: José Luis Ferreiro (president), Pablo Avanzas (secretary), Rut Andrea, Araceli Boraita, David Calvo, Raquel Campuzano, Victoria Delgado, Laura Dos Subirá, Juan José Gómez Doblas, María Antonia Martínez Momblan, Pilar Mazón, Domingo Pascual Figal, Juan Sanchis, José María de la Torre Hernández, David Vivas.
SEC Working Group for the 2023 ESC guidelinesfor the management of cardiomyopathies: Eduardo Villacorta-Argüelles (coordinator), Laura Dos-Subirà (coordinator), Francisco José Bermúdez-Jiménez, Esther González-López, José María Larrañaga-Moreira, Javier Limeres-Freire, Lidia López-García, Carmen Muñoz-Esparza, Esther Zorio-Grima.
* Corresponding author.
The names of all the authors of the article are listed in alphabetical order in Appendix A.
E-mail addresses: evillacorta@secardiologia.es (E. Villacorta Argüelles); laura.dos@vallhebron.cat (L. Dos Subirà).