Tanto las complicaciones macrovasculares como las microvasculares son causas importantes de morbimortalidad en la diabetes tipo 1 y tipo 21, pero hay un aumento de la incidencia de las complicaciones macrovasculares incluso antes del inicio de la diabetes tipo 22. Aunque la glucemia elevada3 y las modificaciones de proteínas y lípidos inducidas por la glucosa (productos terminales de glucación avanzada4) pueden ser desencadenantes de la enfermedad tanto macrovascular como microvascular una vez aparecida la diabetes (tanto de tipo 1 como de tipo 2), desde hace mucho tiempo hay un debate respecto a los factores que causan la enfermedad macrovascular en el contexto del síndrome metabólico y la prediabetes. Ciertamente en la diabetes, y probablemente también en el contexto del síndrome metabólico5, 6, 7, 8, la enfermedad vascular y la enfermedad coronaria ateroscleróticas se producen en mayor medida de lo que explica la acumulación de otros factores de riesgo asociados, como la hipertrigliceridemia, las bajas concentraciones de lipoproteínas de alta densidad y la hipertensión. La resistencia a la insulina antes del inicio de la diabetes se caracteriza, por definición, por la hiperinsulinemia, y desde hace tiempo se ha especulado con la posibilidad de que esto tenga una relación causal con la enfermedad vascular9, 10, 11, 12. En esta breve revisión abordaremos la plausibilidad biológica y la evidencia existente respecto a que la hiperinsulinemia pueda ser un mecanismo causal en el desarrollo de la aterosclerosis antes y después del inicio de la diabetes tipo 2.
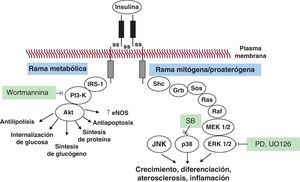
Resistencia selectiva a la insulina e hiperinsulinemia compensatoria: fisiopatologíaInicialmente, Reaven et al definieron el síndrome de resistencia a la insulina como una agrupación de factores de riesgo cardiovascular, que incluía intolerancia a la glucosa, dislipemia e hipertensión, relacionada con una potenciación de la enfermedad cardiovascular13. Reaven et al definieron el síndrome metabólico como un trastorno clínico caracterizado por resistencia a la insulina, deterioro de la glucemia basal en ayunas, obesidad, dislipemia e hipertensión13. Posteriormente se han propuesto otras dos definiciones del síndrome metabólico, una del National Cholesterol Education Program Adult Treatment Panel III14 y la otra de la Organización Mundial de la Salud15. La resistencia a la insulina (medida con el método de referencia del clamp euglucémico hiperinsulinémico o con métodos indirectos, como la prueba de tolerancia a glucosa intravenosa con muestras frecuentes, la prueba de supresión de insulina o el índice HOMA) puede demostrarse en hasta un 76% de los individuos16 y se acompaña de una hiperinsulinemia compensatoria16. Aunque el conocimiento de los mecanismos moleculares de resistencia a la insulina es todavía incompleto, se han descrito anomalías de la señalización de la insulina17. En los tejidos periféricos, incluidos el músculo esquelético y el hígado, en condiciones normales, la insulina inicia su acción uniéndose a su receptor específico de la superficie celular, es decir, el receptor de insulina (IR), que es una proteína heterotetramérica formada por dos subunidades α extracelulares y dos subunidades β transmembrana, conectadas por puentes disulfuro. La unión de la insulina a la subunidad α extracelular induce cambios de conformación del IR que causan, a su vez, la dimerización de los receptores adyacentes y la activación del dominio de tirosincinasa de la parte intracelular de la subunidad β. El inicio de la actividad de tirosincinasa del IR fomenta la autofosforilación de la propia subunidad β y la fosforilación rápida de las denominadas «proteínas de acoplamiento» (docking), como los sustratos de IR (IRS) -1, -2, -3 y -4, y otras varias proteínas, incluidas las proteínas de homología de colágeno (shc) y la homología de SRC 2 (SH2), que activan, a su vez, múltiples sustancias intermedias de señalización intracelular (Figura 1). Así pues, las proteínas IRS, shc y SH2 desempeñan un papel regulador importante en la cascada de señalización de la insulina. En su forma fosforilada, estas proteínas se convierten en puntos de anclaje para las proteínas intracelulares que contienen dominios SH2 complementarios. Concretamente, la interacción entre las proteínas IRS-1 y la fosfatidilinositol (IP) 3-cinasa determina la activación de Akt (también denominada proteincinasa B), que desempeña un papel crucial en el mecanismo de acción de la insulina para la translocación de GLUT-4, el transporte de glucosa y la activación de la óxido nítrico (NO) sintasa («vía de señalización metabólica»). En cambio, los efectos no metabólicos, proliferativos, mitógenos y proinflamatorios de la insulina se producen mediante la activación de la Ras (principalmente a través de shc y, en menor grado, de proteínas IRS), la Raf y las cinasas de proteína activada por mitógenos (MAPK) («vía de señalización de crecimiento»)18. En los animales insulinorresistentes y en modelos in vitro, se puede demostrar una reducción de la activación de la señalización de la insulina a través de la vía de la IRS-1/PI3-cinasa, que da lugar a una disminución de la captación de glucosa, reducción de la síntesis de NO y reducción de la utilización de glucosa en los tejidos diana de la insulina. La misma reducción del transporte de glucosa se percibe en las células beta pancreáticas, e induce un aumento compensatorio de la secreción de insulina. Sin embargo, al mismo tiempo, la vía de la insulina a través de la MAPK se mantiene inalterada19. Es fácil comprender que este desequilibrio selectivo de las dos vías de transducción de señal en situaciones como la hiperinsulinemia puede conducir a una señal excesiva de proliferación/fomento del crecimiento, y al mismo tiempo permitir que se mantengan normales el transporte y la homeostasis de la glucosa. La hiperinsulinemia compensatoria estimula diversos fenómenos proliferativos y proaterogénicos en las células endoteliales y del músculo liso vascular. Estos efectos incluyen un aumento de la producción de inhibidor de activador de plasminógeno tipo 1 (PAI-1), endotelina, citocinas proinflamatorias y un aumento de la expresión de las moléculas de adhesión19, 20, 21, 22.

Figura 1. La vía de señalización de la insulina y su deterioro en la resistencia a la insulina. Tras la unión a su receptor de tirosincinasa, la insulina induce la dimerización del receptor y la activación de una cascada de fenómenos de fosforilación que produce dos clases de efectos: a) efectos «metabólicos», que fomentan el transporte de glucosa, la síntesis de glucógeno y proteínas, la inhibición de la lipolisis, la protección contra la apoptosis y la liberación de óxido nítrico (que se describen de manera amplia como efectos «antiinflamatorios»), y b) efectos de fomento del crecimiento y la diferenciación que conducen a fomento de la inflamación y la aterogénesis (es decir, la señalización de la insulina proinflamatoria y mitógena). Akt: proteincinasa B; eNOS: óxido nítrico sintasa endotelial; ERK: cinasa receptora extracelular; IRS-1: receptor de sustrato de insulina 1; JNK: cinasa terminal c-Jun NH2-1; MEK: proteincinasa activada por mitógenos/cinasa receptora extracelular; p38: proteincinasa activada por mitógenos p38; PD (PD98059) y UO126: inhibidores de cinasa receptora extracelular 1/2; PI3-cinasa: fosfatidilinositol(IP)3 cinasa; wortmannina: inhibidor de PI3-cinasa.
La insulina desempeña un papel importante en el mantenimiento de la homeostasis de los vasos sanguíneos a través de la activación del NO derivado del endotelio. La insulina aumenta la producción de NO endotelial al activar la NOS-III (NOS endotelial) mediante mecanismos postraduccionales rápidos que actúan a través de la vía de señalización de PI3K/Akt23. En los estados de resistencia a la insulina, la vía de PI3K/Akt es inhibida de manera selectiva, y ello lleva a una disfunción endotelial, con el consiguiente aumento del tono vascular e hipertensión, un aumento de la interacción entre células endoteliales y leucocitos y un estado protrombótico. Esta resistencia a la insulina «selectiva» se ha puesto de manifiesto en el músculo esquelético de personas obesas y de pacientes con diabetes tipo 224, así como en los vasos sanguíneos y el miocardio de ratas Zucker obesas. En esta situación, los efectos antiaterogénicos fisiológicos normales de la insulina, debido en gran parte a su capacidad de aumentar la producción de NO, se convierten en efectos proaterogénicos25.
Un círculo vicioso entre hiperinsulinemia y resistencia a la insulinaLas concentraciones plasmáticas elevadas de insulina en estados de resistencia a la insulina pueden desencadenar también un círculo vicioso que aumente aún más la resistencia a la insulina26 mediante la supresión de los efectos que se producen a través del eje PI3K/AKT/NO, y ello puede desequilibrar el sistema como consecuencia de un fomento neto de los efectos relacionados con la activación de la MAPK. Dado que la insulina desencadena una serie de efectos biológicos a través de la unión y activación de su receptor (IR), dotado de actividad de tirosincinasa sobre sustratos específicos, como IRS −1 y −227, los ratones con una deleción específica de los genes de IRS-1 e IRS-2 muestran un fenotipo de resistencia a la insulina28.
En los modelos animales de hiperinsulinemia, como los ratones ob/ob y las ratas Zucker obesas, se observan valores bajos de proteínas IRS-1 e IRS-2 en el hígado29, 30. Estos modelos se caracterizan por tener resistencia a la insulina y reducción de la función del eje IR/IRS-1/PI3K/AKT en el hígado y el músculo esquelético. Se ha demostrado que las incubaciones breves in vitro de mioblastos con concentraciones elevadas de insulina determinan una reducción, mediada por la PI3K, de la expresión de la proteína IRS-1 y una desensibilización de los mecanismos de transducción de la señal de insulina9. Por último, una exposición prolongada de mioblastos en cultivo a concentraciones elevadas de insulina se asocia a una reducción de la actividad del eje IR/IRS-1/PI3K/AKT31. Nosotros hemos demostrado que la exposición prolongada de células endoteliales de vena umbilical humana a concentraciones de insulina elevadas induce una regulación negativa del eje PI3K/AKT/eNOS, que es paralela al aumento de expresión de la molécula de adhesión celular vascular 1 (VCAM-1)32. Sin embargo, los mecanismos moleculares por los que la hiperinsulinemia da lugar a resistencia a la insulina o la agrava son todavía en gran parte desconocidos.
Hiperinsulinemia y enfermedad vascular: evidencia procedente del laboratorioLos experimentos realizados en animales33, 34 y varios estudios in vitro han aportado evidencias de plausibilidad biológica de la hipótesis según la cual las concentraciones altas de insulina son proaterogénicas. La relación entre enfermedad coronaria y concentraciones elevadas de insulina se propuso por primera vez a finales de los años sesenta10 y se confirmó después (véase la revisión de Reddy et al35). In vitro, se ha demostrado que la insulina estimula la proliferación y la migración de las células de músculo liso arterial en preparados tisulares21 e induce la adhesión monocitaria al aumentar la expresión de la VCAM-1 en las células endoteliales22, 36, 37. La VCAM-1 probablemente sea la molécula de adhesión más relevante para el desarrollo de la aterosclerosis38. Este aumento de la expresión en presencia de insulina se produce en un sistema en el que la insulina puede elevar todavía la biodisponibilidad de NO, lo cual inhibiría normalmente la activación endotelial y la aterogénesis39. En consecuencia, estas observaciones indican que el efecto neto de las concentraciones altas de insulina para las células endoteliales es principalmente un fenotipo proinflamatorio. Nosotros hemos demostrado también que estos efectos pueden ser potenciados por el inhibidor de la IP-3-cinasa wortmannina22, lo cual nos lleva a proponer que pueden amplificarse en mayor medida en situaciones de resistencia a la insulina simuladas por la wortmannina. Dado que la capacidad de la insulina de inducir la activación endotelial (para la cual la expresión de VCAM-1 es a la vez un marcador y un mediador) es una explicación plausible de la enfermedad macrovascular que acompaña a los trastornos hiperinsulinémicos, hemos examinado los posibles mecanismos moleculares involucrados en este patrón específico de activación endotelial. Se incubaron células endoteliales de vena umbilical humana con insulina (0-24 h) ± inhibidores de las vías de señalización potencialmente involucradas. La incubación de células endoteliales con inhibidores de ERK1/2 no influyó en la expresión de VCAM-1 inducida por la insulina. En cambio, los inhibidores de p38 MAPK, SB203580 y SB202190, el inhibidor de la isoforma de proteincinasa C (PKC)-β LY379196, y (parcialmente) el inhibidor de la c-Jun terminal NH2 cinasa SP600127, todos ellos evaluados a concentraciones próximas a su media concentración inhibitoria máxima (CI50) para la inhibición de la fosforilación del sustrato, redujeron el efecto de la insulina en la VCAM-1. La silenciación génica de la p38 MAPK por la acción de moléculas pequeñas de ARN de interferencia, que inhibían la expresión de la p38 MAPK, causó una supresión de la expresión de VCAM-1 estimulada por la insulina22, 36, 37. El tratamiento con insulina condujo también a una activación de NF-κB22, 36.
En animales, se ha demostrado que el tratamiento a largo plazo con insulina induce lesiones arteriales que son ricas en lípidos y estimulan el engrosamiento de la pared10. Los mecanismos que causan estas lesiones son el aumento de la síntesis de colesterol en el tejido adiposo, un desequilibrio en la proporción de receptores de lipoproteínas de baja densidad y lipoproteínas de alta densidad (con un aumento de los primeros y una reducción de los segundos) y un aumento de la unión de las lipoproteínas de baja densidad a las células de músculo liso arteriales10. La insulina es también un factor de crecimiento capaz de fomentar la angiogénesis y la proliferación de células de músculo liso mediante la activación de las mismas vías que son activadas por los factores de crecimiento insulínico (IGF)40. Estos efectos de la insulina parecen intervenir en la neovascularización retiniana y desempeñan, pues, un papel clave en la fisiopatología de la microangiopatía diabética y (potencialmente) de la desestabilización de la placa aterosclerótica41, 42, 43.
Otros posibles mecanismos relevantes por los que la elevada concentración de insulina favorece la aterosclerosis son la disfunción endotelial44 y la inhibición de la apoptosis de los macrófagos45. La disfunción endotelial precede a los episodios macrovasculares y los predice. En humanos sanos, la infusión de insulina, al alcanzar unas concentraciones fisiopatológicamente relevantes (> 120pmol/l), puede inducir una disfunción endotelial grave en las arterias grandes44. Los mecanismos probablemente incluyan un aumento del estrés oxidativo intracelular46. Los estudios realizados in vitro han puesto de manifiesto que la insulina estimula la producción de endotelina, la actividad del sistema simpático y la retención de sodio47. Además, la insulina facilita la migración y la proliferación de las células de músculo liso, aumenta la producción de matriz extracelular e induce un estado procoagulante48, con lo que posiblemente contribuye a la reestenosis tras la angioplastia, que se observa con mayor frecuencia en los pacientes diabéticos que en los no diabéticos49.
Hiperinsulinemia y enfermedad cardiovascular: evidencia obtenida a la cabecera del pacienteA pesar de la clara evidencia fisiopatológica y experimental de un efecto proaterogénico de la hiperinsulinemia secundaria a una resistencia a la insulina, es muy frecuente que a los pacientes con diabetes tipo 2 se les administre insulina para normalizar la hiperglucemia, las concentraciones de ácidos grasos libres y la glucohemoglobina. Este tratamiento implica a menudo la administración de insulina a dosis muy altas (de hasta 100 o incluso 625 U/día)50, lo cual causa la aparición de efectos indeseables, como aumento de peso, inhibición de la secreción endógena residual de insulina51 y sobreexpresión de la vía de MAPK19. Sin embargo, dados los efectos favorables de la insulina en la glucemia y los efectos nocivos causados por la glucosa elevada en la función vascular, la evidencia respecto al efecto negativo neto de las dosis altas de insulina en la diabetes no está clara. El estudio DAI (Grupo de Estudio de Diabetes e Informática, Asociación Italiana de Diabetólogos e Instituto Nacional de la Salud de Italia)52, un estudio de cohorte multicéntrico sobre la prevalencia y la incidencia de los eventos cardiovasculares (infarto de miocardio, tromboembolia cerebral y amputaciones periféricas) en pacientes con diabetes tipo 2, puso de relieve que, en comparación con el tratamiento con antidiabéticos orales (como metformina, que no comporta un aumento de la secreción de insulina), el tratamiento con insulina se asoció a un mayor número de eventos cardiovasculares en los varones y las mujeres con diabetes tipo 2. En los pacientes con diabetes tipo 2, se ha demostrado que el tratamiento con insulina aumenta de manera independiente el riesgo de úlceras del pie53, hipertensión54 y la agregación plaquetaria dependiente de adenosin difosfato55. En el Framingham Heart Study, los pacientes diabéticos tratados con insulina fueron los que presentaron mayor incidencia de morbilidad y mortalidad por enfermedad cardiovascular56. En la First National Health and Nutrition Examination Survey, en un total de 7.381 pacientes observados, los que tenían su diabetes tratada con insulina presentaron un aumento del riesgo de muerte por todas las causas y de muerte atribuible a enfermedad cardiovascular57. En el Veterans Affairs Cooperative Study on Glycemic Control and Complications in Type II Diabetes, los pacientes que recibían un tratamiento intensivo con insulina presentaron una incidencia del 32% de eventos cardiovasculares, en comparación con la del 21% en los pacientes que recibían un tratamiento de insulina estándar58. En el estudio Atherosclerotic Risk in Communities59, los pacientes tratados con sulfonilureas (que también aumentan las concentraciones de insulina) presentaron un riesgo relativo de enfermedad cardiovascular de 1,82, mientras que los pacientes tratados con insulina tuvieron un riesgo relativo de 2,64. El estudio de Kumamoto60, en el que los pacientes tratados con insulina no presentaron un aumento del riesgo de enfermedad macrovascular, no hizo una aportación sustancial para abordar esta cuestión, ya que los pacientes eran hipoinsulinémicos y no tenían obesidad. Un estudio reciente61 ha demostrado que la media de amplitud de las desviaciones de la glucemia observadas en los datos de monitorización continua de la glucosa tenía una correlación positiva e independiente con la excreción urinaria de 8-iso-prostaglandina F2α, un marcador del estrés oxidativo, en pacientes con diabetes mal controlada con fármacos hipoglucemiantes orales. Los autores no observaron este tipo de asociaciones en los pacientes con diabetes tipo 1 o tipo 2 tratados con insulina, lo cual indica que el tratamiento insulínico en sí inhibe el estrés oxidativo en esos pacientes. Sin embargo, los efectos de la insulina en la homeostasis celular podrían depender también de las concentraciones de insulina, dado que se ha demostrado que las dosis suprafisiológicas de insulina inducen la generación de especies moleculares de oxígeno reactivo in vitro62. En conjunto, la insulina exógena produce efectos favorables (reducción de la hiperglucemia) y adversos (fomento de la aterogénesis)63. Esto debe ser una advertencia para un uso menos amplio de la insulina en la diabetes tipo 2. En los pacientes con glucemia > 300 mg/dl, una administración inicial de insulina puede reducir la glucotoxicidad50, 64, 65; después de ello, la reducción de la resistencia a la insulina mediante reducción del peso, aumento del ejercicio físico y uso de sensibilizadores a la insulina, como metformina o las glitazonas, probablemente sea una opción más lógica para prevenir las complicaciones cardiovasculares en los pacientes con diabetes tipo 2. Es de destacar que cinco grandes estudios aleatorizados del control intensivo de la glucosa en comparación con el tratamiento estándar en la diabetes tipo 2 no han mostrado reducción alguna de la mortalidad total o de causa cardiovascular58, 66, 67, 68, 69; en cambio, esta reducción sí se ha observado en el estudio EDIC70 en la diabetes tipo 1, en la que la resistencia a la insulina no es el problema principal y el tratamiento insulínico reemplaza el fallo primario de la producción de insulina por las células beta pancreáticas.
ConclusionesLas concentraciones fisiopatológicas de insulina aumentan la producción de endotelina, citocinas proinflamatorias, moléculas de adhesión leucocitaria endotelial y PAI-1 con lo que causan unos efectos proinflamatorios vasculares en general. Los resultados de los estudios realizados in vitro e in vivo apuntan a un papel patogénico de las concentraciones patofisiológicas y farmacológicas de insulina en la enfermedad vascular. Serán necesarias nuevas investigaciones sobre el uso de inhibidores específicos de las vías de MAPK y PKC, como nuevos agentes farmacológicos para abordar la señalización de la insulina proaterogénica.
FinanciaciónEl trabajo original de los autores que se describe aquí fue financiado por subvenciones del Istituto Nazionale per le Ricerche Cardiovascolari a Raffaele De Caterina.
Conflicto de interesesNinguno.
Full English text available from: www.revespcardiol.org
Autor para correspondencia: Istituto di Cardiologia, Università G. d’Annunzio-Chieti, C/o Ospedale SS. Annunziata, Via dei Vestini, 66013 Chieti, Italia. rdecater@unich.it