To the Editor,
Cardiac disease must be systematically evoked in the event of sudden death in the child. Histiocytoid cardiomyopathy (HC) is a rare disease responsible for severe ventricular arrhythmia very early in life. We report 2 cases of HC diagnosed after sudden death in young girls.
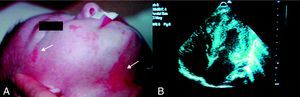
Case 1M. was the first child of healthy, non-blood relative, Caucasian parents. Hypertrophic cardiomyopathy was diagnosed at third trimester by antenatal ultrasound. At birth, clinical examination revealed a systolic cardiac murmur, an axial hypotonia, linear cutaneous erythematous lesions of the face and the neck typical of the microphthalmia with linear skin defects (MLS) syndrome (Figure 1). The EKG registered Wolff-Parkinson-White syndrome. Transthoracic echocardiography demonstrated a biventricular hypertrophic cardiomyopathy, a small perimembranous ventricular septal defect. At 7days of life, she presented an orthodromic tachycardia, successfully treated by amiodarone. Etiologic study of hypertrophic cardiomyopathy was noncontributive. At the age of 3 months, she presented ventricular fibrillation with no resumption of electric activity despite defibrillations. The autopsy confirmed cardiac hypertrophy with micronodules on the mitral and tricuspid valves. Histological analysis revealed areas formed from bundles of myocardial fibers of normal appearance, contrasting with cells of histiocytoid appearance located in the myocardium, the pericardium, and the valves, leading to the diagnosis of HC.

Figure 1. A: linear cutaneous erythematous lesions of the face and the neck (white arrows) typical of the microphthalmia with linear skin defects syndrome. B: trans-thoracic echocardiography showing hypertrophic cardiomyopathy.
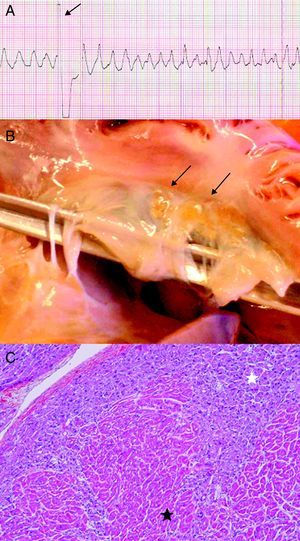
Case 2M. was the first healthy child of non-blood relative, Caucasian parents. At the age of 20 months, she presented a cardiorespiratory arrest secondary to a polymorphic ventricular tachycardia refractory to cardiac defibrillations (Figure 2A). At the autopsy, small yellow nodules were observed on the leaflets of the tricuspid valve (Figure 2B). These nodules were composed of large, foamy, granular cells in the subendocardium. Immunostaining showed perimembranous reactivity for muscle-specific actin, but not for the histiocytic markers (PS100, CD68). Histological analysis revealed the presence of cellular clusters of histiocytoid cells in the subendocardial region, from the apex to the atrium, in the thickness of atrial septum and atrioventricular valve leaflets, resulting in the diagnosis of HC (Figure 2C).

Figure 2. A: polymorphic ventricular tachycardia refractory to cardiac defibrillation. B: yellow arrhythmogenic histiocytoid nodules on leaflets of tricuspid valve (black arrows). C: histological section of the left ventricle displaying large foamy granular cells (white star) and normal myocardic cells (black star).
The diagnosis of HC should be systematically evoked in case of sudden death or severe ventricular arrhythmia in children, mainly in girls less than 2years old.1 Of the reported cases of HC, 19% appeared as sudden deaths in children with no particular history. These patients frequently presented a supraventricular or ventricular arrhythmia uncontrolled by conventional antiarrhythmic treatments. The Wolff-Parkinson-White syndrome, found in almost 10% of the cases, may suggest a diffuse heart disease and may be a marker of gravity. The echocardiographic phenotype is variable. Symmetric or asymmetric hypertrophic cardiomyopathy with a trabeculated endocardium is very suggestive. Cardiac malformations (ventricular septal defect, atrial septal defect, Shone syndrome) are reported. A normal echocardiography may be found in a focal form of the disease. Histologically, HC is characterized by the presence of nodules with more or less scattering in the myocardium or the endocardium.2 The excision of arrhythmogenic nodules is the only treatment that may produce a complete cure and disappearance of arrhythmia.3 Histiocytoid cells have an arrhythmogenic potential and conventional treatment of arrhythmia is not effective. In every case, medical treatment resulted in the death of the patient. For the rare patients who benefit from surgical excision, the long-term prognosis is unknown. Heart transplantation was reported once, in a 30-month-old child.4 The etiopathogenesis of the HC is not yet clearly established; HC should be considered as an X-linked mitochondrial disease affecting cardiac muscle. This hypothesis seems confirmed following an association with MLS syndrome, a rare neurodevelopmental disorder that associates linear cutaneous erythematosus lesions of the face and the neck, eye abnormalities, and neurological lesions in females monosomic for Xp22.3.5
Corresponding author: labombarda-f@chu-caen.fr