To the Editor,
We read with interest the editorial by Drs. Freedman and Iafrati1 recently published in Revista Española de Cardiología. The editorial mentions that prasugrel is an inhibitor of the P2Y12 receptor, that it is not metabolized in the liver, and that it does not appear to be affected by the variability of isozyme P450. We believe that it might be a good idea here to indicate the pharmacokinetic characteristics of prasugrel.
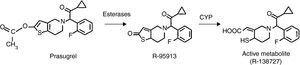
Prasugrel (CS-747, LY-640315) is a third generation, orally administered thienopyridine that acts as a specific and irreversible antagonist of the 5’-diphosphate (ADP) P2Y12 receptor, and which needs to be metabolized for it to exert its effect.2, 3, 4 The initial molecule, prasugrel, is rapidly hydrolyzed by intestinal and blood esterases to the metabolite thiolactone (R-95913) (Figure 1). Thus, prasugrel is not detected in the plasma. Via the action of cytochrome P450 (CYP), this intermediate metabolite is turned into the active metabolite R-138727, which bonds covalently and irreversibly to receptor P2Y12.2, 3

Figure 1. Structural changes required for the activation of prasugrel.
This active metabolite of prasugrel reaches its peak in the plasma after about 30 min and in a manner proportional to the dose given (between 5 mg and 60 mg). When it does not bind to the platelets its half life is about 7h. It should be remembered that the CYP enzymes involved in the metabolism of clopidogrel and prasugrel are polymorphic, ie, they differ between individuals, which partly accounts for the wide variation seen between patients in their response to clopidogrel.3, 4
The metabolism of prasugrel differs from that of clopidogrel in that the metabolism of the latter renders inactive close to 85% of the drug absorbed and two passes through the liver are required (CYP), which influences the variation in individual response.2 In contrast, prasugrel is more efficiently converted into its active metabolite via a process of hydrolysis led by carboxyesterases (mainly intestinal), followed by a single pass through the hepatic CYP (3A4, 2B6, 2C9, 2C19) step. This partly explains its greater bioavailability and more efficient antiaggregant effect compared to clopidogrel.5
Corresponding author: carlosfbarrera@yahoo.com