To the Editor:
Genetic studies have recently identified a novel disease caused by mutations in the X-linked lysosome-associated membrane protein-2 (LAMP2) gene: Danon disease. We report the noninvasive characterization of a patient with this pathology.
A 19-year-old man was admitted to hospital for the evaluation of new onset of heart failure. There was a 4-week history of gradually progressing exercise intolerance. On admission, the patient presented in New York Heart Association class II. His mother died at the age of 44 years with suspicion of dilated cardiomyopathy. On physical examination, cachectic appearance was noted. Auscultation revealed bilateral rales halfway up and hepatomegaly. Routine blood tests produced elevated troponins, creatine kinase and liver enzyme levels. The electrocardiogram was consistent with left bundle branch block and high QRS voltage. A chest X-ray showed cardiomegaly with massive pulmonary congestion.
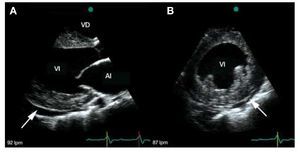
Transthoracic echocardiography demonstrated biventricular symmetrical concentric hypertrophy without left and right outflow tract gradient. The intraventricular septum and posterior wall thickness were 25 mm and 24 mm, respectively. The echogenicity of the whole myocardium was evenly increased (Figure 1a,b). The left ventricle (LV) was dilated, with an end-diastolic diameter of 69 mm, and both atria were enlarged. The systolic function was decreased due to global hypokinesis with akinetic apical segment. Ejection fraction assessed by Simpson's method was 27%. Right ventricular function was impaired, with free wall hypokinesis (Tricuspid Annular Plane Systolic Excursion [TAPSE] - 11 mm). There were signs of pulmonary hypertension (tricuspid regurgitation gradient 50 mm Hg) and moderate pericardial effusion (circumferential, maximum 8.5 mm laterally). Doppler echocardiography showed restrictive transmitral flow pattern.

Figure 1. A: transthoracic echocardiographic parasternal long-axis view demonstrating moderate hypertrophy, increased echogenicity of the myocardium, enlarged left ventricle cavity, left atrial dilatation and pericardial effusion (arrow). B: transthoracic echocardiographic parasternal short-axis view showing concentric left ventricular thickening with a pericardial effusion (arrow). The echogenicity of the myocardium is increased.
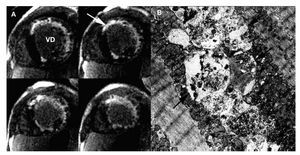
In a cardiovascular magnetic resonance study, perfusion defects, mainly subendocardial, were visible in almost all segments on first pass images acquired at rest. Late gadolinium enhancement was present in the subendocardium and transmurally in the anterior and lateral walls in a distribution that would be unusual in more common pathologies, including amyloidosis and Anderson-Fabry disease (Figure 2a). Endomyocardial biopsy of the right ventricle and skeletal muscle biopsy were obtained. Electron microscopic examination demonstrated the presence of characteristic vacuolar myopathy with vacuoles containing glycogen and cytoplasmic degradation products (Figure 2b). The LAMP2 deficiency in immunofluorescence study enables confirmation of the diagnosis of Danon disease. The patient died of heart failure within 2 months while waiting for cardiac transplant.

Figure 2. A: late-enhancement sequences of the MR study in short axis orientation showing late gadolinium enhancement (arrow) in the subencocardium and the anterior and lateral wall. B: electron microscopic analysis demonstrating autophagic vacuoles (arrows) located within intramyofibrillar space. x 1000.
Danon disease, a rare X-linked dominant disorder, is a lysosomal glycogen storage disease related to mutations in the gene encoding LAMP2 that lead to marked deficiency or complete absence of this protein. LAMP2 mutations typically cause multisystem glycogen storage disease but can also present as primary cardiomyopathy, as in our patient. The histopathologic hallmarks are intracytoplasmic vacuoles containing autophagic material and glycogen in skeletal and cardiac muscle cells.1 This case is one of only a few reports concerning the echocardiographic findings in patients with Danon disease. Previously described echocardiographic features associated with Danon disease mimic HCM due to sarcomere proteins mutations.2,3 Only the average maximal left ventricular wall thickness was characterized as greater than that typically found in patients with HCM (35+/-15 mm)2. In our patient, hypertrophy was smaller (maximal left ventricular wall thickness was 25 mm).
Given the poor prognosis associated with cardiomyopathy due to LAMP2 mutation, early diagnosis is critical for determining the appropriate treatment strategy and achieving timely cardiac transplantation, the only effective therapeutic option.