Myocardial interstitial fibrosis is a constant pathological finding in structural heart diseases of various etiologies that evolve with heart failure. Although fibrosis facilitates heart failure progression, until now no therapeutic strategy has been developed that ensures its reversal. A possible explanation for this may lie in the vision of myocardial interstitial fibrosis as a homogeneous lesion instead of a heterogeneous lesion in which different phenotypes can be distinguished using appropriate criteria. In addition, the notion that the heterogeneity of myocardial interstitial fibrosis may be cardiac disease-specific must be also considered when approaching this entity. Therefore, we propose that myocardial interstitial fibrosis represents a true challenge for transitioning from usual care to biomarker-based personalized treatment and precision medicine in heart failure. As a proof-of-concept, in this review we discuss the phenotyping of myocardial interstitial fibrosis in patients with heart failure attributable to hypertensive heart disease based on its histomolecular alterations and provide evidence of the prognostic relevance of the resulting stratification. Furthermore, we discuss the available information on some circulating biomarkers and certain pharmacological agents useful for noninvasive identification and personalized treatment, respectively, of those phenotypes.
Keywords
A principle of precision medicine in cardiology is to establish the precise phenotype for any given cardiac disorder by means of, among other methodologies, the identification of biomarkers that characterize phenotypic heterogeneity to help diagnose, aid prognosis, and dictate optimal treatment in specific unmet clinical needs.1 In this dual conceptual and methodological framework, it has been proposed that the development and implementation of biomarker-based precision medicine in heart failure (HF) offers the potential to significantly reduce the clinical and financial burdens associated with this syndrome.2 Accordingly, a broad research agenda is currently ongoing that aims to identify targetable phenotypes of HF with biomarkers.3,4 In particular, a new view is emerging that beyond classic biomarkers of myocardial stress/injury, neurohormonal activation and comorbidities, biomarkers that identify targetable phenotypes of the pathological structural remodeling of the failing myocardium may be useful to enhance traditional methods of assessing HF patients,5 as it is well understood that the characteristics of myocardial remodeling may vary depending on the etiology of HF.
For instance, in conditions of chronic hypertension leading to hypertensive heart disease (HHD), the histological components of the myocardium undergo a series of lesions (ie, cardiomyocyte growth and death, interstitial inflammation and fibrosis, reduced maximal cross-sectional area of prearterioles and arterioles, decreased capillary density, and rarefaction of lymphatic vessels) that are governed by a complex interplay of mechanisms.6 In particular, diffuse myocardial interstitial fibrosis (MIF) not only contributes to the progressive impairment of cardiac function, thus facilitating HF, but also adversely influences clinical outcome in patients with HF due to HHD.7 Of note, pharmacological regression of MIF and improvement in cardiac function has been demonstrated in hypertensive rodents of diverse etiologic origins and hypertensive humans,8 supporting the view that the reversal of this lesion is indeed feasible. Therefore, it has been proposed that MIF phenotyping may potentially improve HF patient prognosis through the personalized diagnosis and treatment of cardiac disease-specific MIF.9
In this article, we discuss emerging data supporting the view that the histomolecular heterogeneity of MIF results in different phenotypes with a variable clinical impact in patients with HF attributable to HHD. In addition, we will also address how the histomolecular phenotypic heterogeneity of MIF in these patients can be captured using noninvasive biomarkers, thereby allowing a tailored therapeutic approach to MIF.
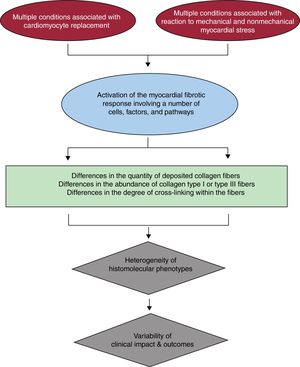
HISTOMOLECULAR HETEROGENEITY OF MIFIn brief, MIF occurs when a number of profibrotic factors acting on diverse cell types present in the heart, namely resident cardiac fibroblasts, promote their transformation into myofibroblasts whose secretome includes factors leading to the predominance of collagen fiber synthesis over its degradation.10 The profibrotic factors may be activated as part of the response that follows the loss of cardiomyocytes (ie, reparative fibrosis) or by mechanical and nonmechanical (eg, neurohormonal, inflammatory, or metabolic) stress to the heart (ie, reactive fibrosis). A growing body of evidence suggests that both the quantity and quality of deposited collagen fibers must be taken into account for a more comprehensive understanding on how the histomolecular heterogeneity of MIF results in the diversity of its clinical impact in patients with HF due to HHD (figure 1).

Quantitatively, MIF is characterized by a diffuse deposition of excess collagen fibers relative to the cardiomyocyte mass. Collagen deposits may appear as microscars and large strands located within the interstitial space surrounding individual cardiomyocytes and cardiomyocyte fascicles or around the intramyocardial vessels. Excess collagen deposition (CD) is detected by an increase in the percentage of total myocardial tissue occupied by collagen fibers determined in myocardial samples stained with collagen-specific stains and analyzed using automated image analysis systems. Interestingly, a strong correlation (r> 0.95) has been reported between CD and the amount of collagen type I fibers, but not of collagen type III fibers, in the myocardium of patients with HF due to HHD,11,12 suggesting that MIF is the result of excessive deposition of collagen type I.
Qualitative changes related to the molecular organization of collagen fibers can also be important regarding the impact of MIF on cardiac function.13 For instance, the degree of intramolecular and intermolecular covalent bonds between the collagen molecules that constitute the collagen fiber (ie, collagen cross-linking [CCL])14 may vary and consequently the physicochemical properties of the fiber may also change (ie, the higher the CCL the lower the solubility of the fiber and the higher its stiffness and resistance to degradation),15 thus impacting the mechanical properties of the myocardium.16 CCL is mediated by enzymatic (lysyl oxidases [LOX-1 and LOX-like enzymes] and transglutaminases) and nonenzymatic (advanced glycation end products) mechanisms.17,18 Interestingly, increased CCL, measured as the ratio of insoluble collagen to soluble collagen in myocardial samples has been found in patients with HF due to HHD exhibiting MIF and increased left ventricular (LV) chamber stiffness.12,19 In addition, myocardial LOX-1 has been found to be increased in these patients, its expression being localized in areas of fibrosis and associated with the degree of CCL, the amount of insoluble collagen and collagen type I fibers, and LV stiffness.19,20 Of interest, these findings were reported in hypertensive patients with HF with either preserved or reduced ejection fraction.
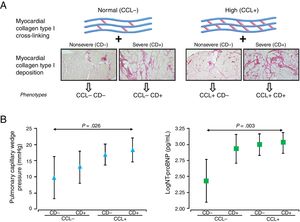
PHENOTYPING THE HISTOMOLECULAR HETEROGENEITY OF MIFThe clinical relevance of the histomolecular heterogeneity of MIF is advanced by findings from observational studies demonstrating that increased CD in HF patients is associated with long-term all-cause mortality,21 and increased CCL is associated with long-term HF hospitalization.22 Therefore, as a proof-of-concept approach, our group recently investigated whether the analysis of both CD and CCL allows diverse phenotypes of MIF to be identified in patients with HF attributable to HHD presenting with variable clinical characteristics and outcomes.23 To this end, patients were first classified according to values below or above CCL reference values as patients with normal CCL (or CCL−) or patients with high CCL (or CCL+), respectively. Similarly, patients were also classified according to values below or above CD reference values as patients with nonsevere CD (or CD−) or patients with severe CD (or CD+), respectively. Then, patients were categorized in 4 subgroups as CCL− CD−, CCL− CD+, CCL+ CD− and CCL+CD+(figure 2A) and the histopathologic and clinical characteristics of the 4 subgroups were compared. The amount of myocardial collagen type I, but not collagen type III, increased significantly in the subgroups. The pulmonary capillary wedge pressure and the amino-terminal propeptide of brain natriuretic peptide also increased significantly in the subgroups and showed significant adjusted differences between the CCL+CD+and CCL− CD− subgroups (figure 2B). No significant differences in the frequency of comorbidities or HF with preserved or reduced ejection fraction were observed among the 4 subgroups. After a median follow-up of 8.15 years, a primary composite outcome of first hospitalization for HF (HHF) after enrolment or death from cardiovascular causes and a secondary composite outcome of first HHF after enrolment or all-cause death increased significantly in the subgroups, with the CCL+CD+bioprofile showing the highest risk.23
 and collagen cross-linking (CCL) as patients with nonsevere CD (CD−) or severe CD (CD+) and patients with normal CCL (CCL−) or high CCL (CCL+). B: symbols represent age- and sex-adjusted means and 95% confidence intervals of pulmonary capillary wedge pressure (triangles) and amino-terminal propeptide of brain natriuretic peptide (NT-proBNP) (log) (squares) in patients with heart failure attributable to hypertensive heart disease classified according to the previously mentioned histomolecular phenotypes of myocardial interstitial fibrosis. Adapted with permission from Ravassa et al.23")
A: classification of patients with heart failure of hypertensive etiology in 4 distinct histomolecular phenotypes of myocardial interstitial fibrosis according to reference values of myocardial collagen deposition (CD) and collagen cross-linking (CCL) as patients with nonsevere CD (CD−) or severe CD (CD+) and patients with normal CCL (CCL−) or high CCL (CCL+). B: symbols represent age- and sex-adjusted means and 95% confidence intervals of pulmonary capillary wedge pressure (triangles) and amino-terminal propeptide of brain natriuretic peptide (NT-proBNP) (log) (squares) in patients with heart failure attributable to hypertensive heart disease classified according to the previously mentioned histomolecular phenotypes of myocardial interstitial fibrosis. Adapted with permission from Ravassa et al.23
These findings show that the histomolecular heterogeneity of MIF in patients with HF of hypertensive etiology translates into a diversity of phenotypes with varying clinical implications, including a high-risk phenotype in approximately 30% of patients in whom the concurrence of severe CD and high CCL is associated with severe LV dysfunction and increased vulnerability to both HF hospitalization and mortality. Whether these phenotypes can be also found in patients with HF due to other etiologies different from HHD deserves further studies.
BIOMARKER-BASED IDENTIFICATION OF THE HISTOMOLECULAR PHENOTYPES OF MIFThe use of biomarkers that reflect the concurrence of severe CD and high CCL in a given patient may help to identify HF patients carrying the high-risk histomolecular phenotype of MIF and thus susceptible to receiving personalized therapies to prevent the underlying collagen alterations and improve their prognosis.
It has been proposed that the measurement of myocardial extracellular volume (ECV) using cardiac magnetic resonance or computed tomography enables the detection of diffuse MIF.24 However, ECV appears to reflect the quantity of collagen fiber deposits (ie, CD) but not their quality (ie, CCL).25 In fact, a recent study performed in intraoperative interventricular septum biopsies from 133 patients with aortic stenosis suggests that ECV is not able to simultaneously capture both of these 2 quantitative and qualitative aspects of fibrosis.26 Therefore, a complementary biomarker-based approach is required to identify the histomolecular phenotypic heterogeneity of MIF in HF.
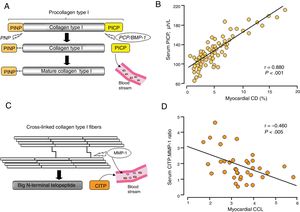
Although the assessment of a number of circulating molecules has been proposed for the noninvasive assessment of MIF,27,28 only a few of them accomplish the criteria to be considered true biomarkers of MIF.29 In this regard, findings from our laboratory suggest that 2 circulating biomarkers related to extracellular collagen type I processing may be useful to assess the histomolecular phenotypes of CD and CCL in patients with HF attributable to HHD. We have demonstrated that serum levels of the procollagen type I carboxy-terminal propeptide (PICP), formed during the conversion of procollagen type I into mature fibril-forming collagen type I by the enzyme procollagen C-proteinase or bone morphogenetic protein-1 (PCP/BMP-1) (figure 3A), are directly correlated with myocardial CD in HF patients (figure 3B).11,12,30 On the other hand, as previously mentioned, the degree of CCL determines the resistance of collagen type I fibers to degradation by matrix metalloproteinase-1 (MMP-1), resulting in diminished cleavage of the carboxy-terminal telopeptide of collagen type I (CITP) (figure 3C). We have recently shown that the serum CITP:MMP-1 ratio is inversely correlated with myocardial CCL in HF patients (figure 3D).22 It has to be taken into account that neither PICP nor the CITP:MMP-1 ratio are cardiac-specific biomarkers despite their associations with myocardial CD and CCL, respectively, and that HF is characterized by the presence of comorbidities also affecting collagen metabolism (eg, chronic kidney disease).31
. B: direct correlation between myocardial collagen deposition (CD) and PICP in patients with heart failure attributable to hypertensive heart disease. Adapted with permission from López et al.30 C: diagrammatic representation of collagen type I cross-linked carboxy-terminal telopeptide (CITP). D: inverse correlation between myocardial collagen cross-linking (CCL) and the serum ratio of CITP and total matrix metalloproteinase-1 (CITP:MMP-1) in patients with heart failure attributable to hypertensive heart disease. Adapted from López et al.22 PCP/BMP-1, procollagen C-proteinase/bone morphogenetic protein-1; PINP, procollagen type I amino-terminal propeptide; PNP, procollagen N-proteinase.")
A: diagrammatic representation of the extracellular origin and destination of fibrillar collagen-derived peptides procollagen type I carboxy-terminal propeptide (PICP). B: direct correlation between myocardial collagen deposition (CD) and PICP in patients with heart failure attributable to hypertensive heart disease. Adapted with permission from López et al.30 C: diagrammatic representation of collagen type I cross-linked carboxy-terminal telopeptide (CITP). D: inverse correlation between myocardial collagen cross-linking (CCL) and the serum ratio of CITP and total matrix metalloproteinase-1 (CITP:MMP-1) in patients with heart failure attributable to hypertensive heart disease. Adapted from López et al.22 PCP/BMP-1, procollagen C-proteinase/bone morphogenetic protein-1; PINP, procollagen type I amino-terminal propeptide; PNP, procollagen N-proteinase.
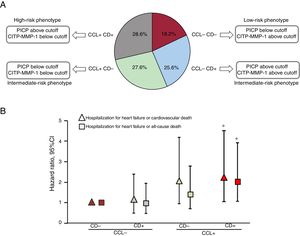
Considering these limitations, we have investigated whether the combination of serum PICP and the CITP:MMP-1 ratio captures the variable clinical outcome of the different CD plus CCL histomolecular patterns in patients with HF attributable to HHD.23 Previously, in patients with both endomyocardial biopsies and blood samples, we identified the cutoff value of PICP for identifying patients with severe CD (CD+), and the CITP:MMP-1 ratio cutoff values for identifying patients with high CCL (CCL+) using receiver operating characteristic curves (figure 4A). With these values, HF patients were categorized in 4 biomarker-based bioprofiles as: low-risk phenotype (CCL− CD−), intermediate-risk phenotypes (CCL− CD+and CCL+CD−) and high-risk phenotype (CCL+CD+) (figure 4A), and the clinical outcomes of the 4 bioprofiles were compared. After a median follow-up of 5.31 years, the composite outcomes HHF or cardiovascular death and HHF or all-cause death significantly increased in the different groups, with significant differences between CCL+CD+and CCL− CD− bioprofiles (figure 4B). Moreover, this classification improved the prognostic performance of important risk factors. Using the same biomarker-based approach, we recently found that the CCL+CD+bioprofile was associated with a higher prevalence of atrial fibrillation (AF), incidence of new-onset AF, and recurrence of AF after pulmonary vein ablation than the other phenotypes in patients with HF attributable to HHD.30 In addition, it improved the predictive value of relevant AF risk factors.32
 or severe collagen deposition (CD+) and patients with normal collagen cross-linking (CCL−) or high collagen cross-linking (CCL+) defined by the values of serum procollagen type I carboxy-terminal propeptide (PICP), and by the values of the serum ratio of collagen type I cross-linked carboxy-terminal telopeptide (CITP) to matrix metalloproteinase-1 (MMP-1) ratio (CITP:MMP-1). B: symbols represent adjusted hazard ratios and 95% confidence intervals for heart failure hospitalization or cardiovascular death (triangles) and heart failure hospitalization or all-cause death (squares) in patients with heart failure attributable to hypertensive heart diseased classified according to the previously mentioned biochemical phenotypes of myocardial interstitial fibrosis. * P <.05 compared with CD− CCL− patients. Adapted with permission from Ravassa et al.23")
A: frequency distribution of the histomolecular phenotypes of myocardial interstitial fibrosis in patients with heart failure attributable to hypertensive heart disease classified as patients with nonsevere collagen deposition (CD−) or severe collagen deposition (CD+) and patients with normal collagen cross-linking (CCL−) or high collagen cross-linking (CCL+) defined by the values of serum procollagen type I carboxy-terminal propeptide (PICP), and by the values of the serum ratio of collagen type I cross-linked carboxy-terminal telopeptide (CITP) to matrix metalloproteinase-1 (MMP-1) ratio (CITP:MMP-1). B: symbols represent adjusted hazard ratios and 95% confidence intervals for heart failure hospitalization or cardiovascular death (triangles) and heart failure hospitalization or all-cause death (squares) in patients with heart failure attributable to hypertensive heart diseased classified according to the previously mentioned biochemical phenotypes of myocardial interstitial fibrosis. * P <.05 compared with CD− CCL− patients. Adapted with permission from Ravassa et al.23
Therefore, the combination of serum PICP and the CITP:MMP-1 ratio is a good reflection of the variable clinical impact of the histomolecular phenotypes of MIF in patients with HF due to HHD. Furthermore, the bioprofile defined by the combination of high serum PICP and low CITP:MMP-1 ratio identifies patients with the high-risk phenotype (CCL+CD+) characterized by increased risk of AF, HHF, and mortality.
TAILORED TREATMENT OF MIF BASED ON THE HISTOMOLECULAR PHENOTYPESFrom a mechanistic point of view, it can be hypothesized that, in patients with the high-risk CD+CCL+MIF phenotype, the fibrogenic PCP/BMP-1−LOX-1 axis predominates over the fibrolytic MMP-1 axis, thus offering an opportunity for tailored antifibrotic therapies in these patients based on the inhibition of the PCP/BMP-1−LOX-1 axis. In this regard, whereas some clinical findings highlight the potential of targeting PCP/BMP-1 and LOX-1 with pharmacological agents currently used to treat HF patients, the identification of novel drugs targeting these molecules is currently ongoing.
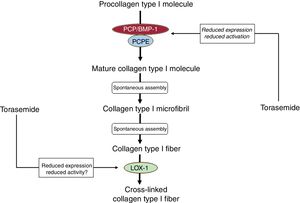
In patients with HF due to HHD, administration of torasemide in addition to standard HF therapy was associated with reductions in myocardial CD and CCL in conjunction with decreased activation of PCP/BMP-1, reduced expression of its enhancer-1 (PCPE-1), and diminished expression of LOX-1.19,30,33 Additionally, torasemide treatment was accompanied by normalization of LV stiffness and improvement of function in 80% of the patients, without LV enlargement.18 None of these effects were observed in furosemide-treated patients.19,30,33
Therefore, beyond its renal tubular actions resulting in increased natriuresis and diuresis, torasemide may exert cardiac antifibrotic actions interfering with the fibrogenic PCP/BMP-1−LOX-1 axis (figure 5). Whether HF patients with the high-risk CCL+CD+phenotype of MIF would benefit more from the antifibrotic properties of torasemide is a hypothesis that remains to be tested in adequately designed trials with patients stratified according to the PICP and CITP:MMP-1 ratio bioprofile.

Proposed inhibitory effects of torasemide on the enzyme-mediated steps of the extracellular process leading to the generation of collagen type I fibers. LOX-1, lysyl oxidase-1; PCP/BMP-1, procollagen C-proteinase/bone morphogenetic protein-1; PCPE-1, procollagen C-proteinase enhancer-1.
Novel antifibrotic compounds based on the modulation of PCP/BMP-1-mediated mature collagen formation and of LOX-1-mediated CCL are currently under development, but their safety and effectiveness in cells in culture and in experimental in vivo models remains to be explored. Of interest, the non-AT1 receptor blocker aldehyde metabolite of losartan EXP3179 has been reported to fully prevent LOX-1, CCL and CD increase without normalization of blood pressure in rats with experimentally induced hypertension and MIF.34 In contrast, administration of the AT1 receptor blocker acid metabolite of losartan, EXP3174, normalized blood pressure and attenuated fibrosis but did not modify LOX-1 and CCL.34 The potential translation of these effects can be anticipated taking into account that losartan administration is associated with blood pressure-independent regression of MIF and reduction of LV stiffness in hypertensive patients.35
CONCLUSIONS AND FUTURE DIRECTIONSMIF is extremely heterogeneous, as several stages of the fibrotic process exist, each with different pathophysiological components resulting in a diverse histomolecular expression and clinical behavior. Detection and therapy of MIF suffer from a lack of precision and thus strategies that allow for differentiation of MIF phenotypes in a disease-specific way are needed, ideally combining bioimaging and circulating biomarkers. In this conceptual framework, here we have reviewed one example of biomarker-based bioprofiling of the histomolecular heterogeneity of MIF in patients with HF attributable to HHD. This approach may allow stratification of patient risk and setting the stage for a personalized treatment, in particular of those patients with the highest risk. It is urgent that academia, industry, and regulators collaborate to develop a broad research agenda (including a randomized trial) investigating whether this and other similar antifibrotic approaches ensure better management and care of patients with HF due to HHD or other etiologies.
FUNDINGThis project was supported by the Ministerio de Ciencia, Innovación y Universidades, Spain (Instituto de Salud Carlos III grants CB16/11/00483 and PI18/01469 co-financed by European Fund for Regional Development funds), the European Commission FP7 Programme (FIBRO-TARGETS project 2013-602904 and HOMAGE project 2012-305507) and the ERA-CVD Joint Transnational Call 2016 LYMIT-DIS (AC16/00020) and LIPCAR-HF (AC16/00016).
CONFLICTS OF INTERESTNone declared.