
Mitochondria are dynamic organelles able to vary their morphology between elongated interconnected mitochondrial networks and fragmented disconnected arrays, through events of mitochondrial fusion and fission, respectively. These events allow the transmission of signaling messengers and exchange of metabolites within the cell. They have also been implicated in a variety of biological processes including embryonic development, metabolism, apoptosis, and autophagy. Although the majority of these studies have been confined to noncardiac cells, emerging evidence suggests that changes in mitochondrial morphology could participate in cardiac development, the response to ischemia-reperfusion injury, heart failure, and diabetes mellitus. In this article, we review how the mitochondrial dynamics are altered in different cardiac pathologies, with special emphasis on heart failure, and how this knowledge may provide new therapeutic targets for treating cardiovascular diseases.
Keywords
Heart failure is a complex condition characterized by the inability of the heart to provide an adequate cardiac output to match the metabolic demands of the remaining tissues. The condition represents the terminal phase of ischemic heart disease, valve disease, and hypertensive conditions.1 In industrialized countries, where the life expectancy of the population and survival after other heart diseases are high, heart failure is a highly prevalent condition.2, 3
An understanding of the pathophysiology of heart failure has enabled standard therapy to be established based on the use of β-blockers in combination with renin-angiotensin-aldosterone system blockade through angiotensin-converting enzyme inhibitors, angiotensin II receptor antagonists, aldosterone receptor antagonists, and in some cases, resynchronization therapy.4 Despite these advances, the annual mortality associated with heart failure remains close to 10%, and it is not known why the disease progresses even with optimal therapy.5 The above has highlighted the need to develop new therapeutic targets based on other abnormal molecular and cellular mechanisms in this disease.
During the 1950s, electron microscopy studies showed that mitochondria are individual double-membrane organelles, whose internal membrane has characteristic folds known as mitochondrial crests.6 This view has been reconsidered in light of recent evidence showing that these organelles form a highly interconnected and dynamic network, whose morphology varies according to cell type.7 As shown in Figure 1A, mitochondria in adult hearts are organized in discrete and packaged structures that are arranged along the myofibrillae. This characteristic distribution is a phenotype acquired during development as the mitochondrial network of the neonate cardiac myocyte (Figure 1B) extends throughout the cytoplasm, distributed differently than in adults, and the morphology is more reminiscent of that observed in other noncardiac cells.8

Figure 1. Mitchondrial morphology of the cardiac myocyte. A, electron microscopy of neonate rat cardiac myocyte in culture (left panel) and adult rat cardiac tissue (right panel); in the neonate cardiac myocyte, the mitochondria (M) are distributed between the cytoplasm and around the nucleus (N), whereas in the adult heart, mitochondria are aligned between the sarcomeric units (S). B, confocal microscopy of live neonate cardiac myocytes treated with the Green MitoTracker probe. The central photograph shows the mitochondrial morphology in control conditions; the transduction of cardiac myocyte with antisense adenovirus for mitofusin protein 2 (left panel) produces fragmentation of the mitochondrial network through decrease in the fusion processes, whereas adenoviral expression of a dominant negative form of the protein related to dynamin-1 (right panel) leads to a reduction in fusion processes.
Recent findings indicate that changes in mitochondrial morphology could be relevant in cardiovascular pathophysiology, as such changes may have an impact on cellular metabolic capacity.9 In this article, we review the current concept of “mitochondrial dynamics” and how this becomes altered in different cardiovascular diseases, with special emphasis on heart failure. In addition, potential new therapeutic targets are highlighted.
CARDIAC METABOLISMIn order to maintain contractile function, the normal adult human heart requires a daily supply of adenosine triphosphate (ATP) equivalent to approximately 30 Kg, that is, around 70 times the weight of the organ.10 Given that the cardiac ATP content is very low (5 μmol/g) and that its hydrolysis rate is very high (approximately 30μmol·g–1·min–1 at rest), all ATP is replenished in approximately 10s, and 95% is synthesized through oxidative phosphorylation.11 In the human adult heart, approximately 70% of cellular ATP is derived from β-oxidation of free fatty acids (FFAs).11
The metabolites generated by β-oxidation and glycolysis are incorporated into the Krebs cycle, and reduced nicotinamide adenine dinucleotide and reduced flavin adenine dinucleotide are generated. These reducing agents are oxidized by the electron transporting chain in the mitochondria, and ATP is generated. Once the chemical energy has been “stored” in ATP, it is transferred to creatine by phosphorylation through a reaction catalyzed by mitochondrial creatine kinase. Phosphocreatine is a smaller molecule than ATP that helps ATP diffusion throughout the contractile apparatus of the cell, where it relinquishes its phosphate load to adenosine diphosphate (ADP) and ATP is regenerated for in situ use. Finally, the creatine returns to the mitochondria to initiate a new cycle.12 At times of high energy demand, the creatine cycle permits ATP generation at a rate 10 times greater than the highest capacity of oxidative phosphorylation.13
Both clinical studies and animal models have demonstrated that in heart failure the heart shows abnormalities in the processing of metabolic substrates, ATP production, and the creatine cycle.14 Patients with cardiac insufficiency have reduced concentrations of phosphocreatine, and this leads to a state of energy depletion that correlates with clinical progression of the disease.14 These findings have prompted the study of cardiac energy metabolism as a new therapeutic target.
MITOCHONDRIAL DYNAMICSMitochondria, in addition to being the compartment for many essential biochemical reactions in situations of energy homeostasis, play a key role in cell death and aging.15 This organelle forms a complex, interconnected, and highly dynamic network that is maintained by the continuous opposing and counterbalanced events of mitochondrial fusion and fission.9, 16 Both the number of tubules and their connections, as well as the subcellular distribution of the organelle, are actively controlled. Thus, the term “mitochondrial dynamics” has been coined to encompass at least 3 different processes: a) remodeling of the mitochondrial network through fusion/fission processes, which is closely linked to the metabolic status of the cell and is controlled by the activity of a group of guanosine triphosphatases (GTPases) related to the dynamin family (Figure 2); b) subcellular mitochondrial motility, particularly relevant in polarized cells and which corresponds to displacement of the mitochondria according to the kinesin 1 and 3 motors and the Milton and Miro adaptors,17 which ensure local supply of ATP in biological processes with high energy requirements and the use of organelles as calcium buffers18, and c) remodeling of the mitochondrial ultrastructure and condensation of its matrix, processes traditionally considered as reflecting the mitochondrial metabolic status.19 Thus, an example of the interconnectivity of the different functional states of the mitochondria and its ultrastructure is the remodeling of the mitochondrial crests observed in transitions between the different respiratory states or during apoptosis.20

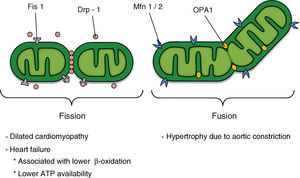
Figure 2. Potential participation of mitochondrial dynamics in cardiovascular disease; the factors that regulate mitochondrial morphology are shown in the upper section of the figure. ATP, adenosine triphosphate; Drp-1, dynamin-related protein-1; Fis1, fission 1 protein 1; Mfn 1/2, mitofusins 1 and 2; OPA1, optical atrophy 1.
THE MACHINERY OF MITOCHONDRIAL FISSIONMitochondrial fission is regulated in mammals by, among other things, the activities of the dynamin-related protein-1 (Drp-1) and fission protein 1 (Fis1). Drp-1 has a sequence homologous with the dynamins, GTPases that regulate vesicular trafficking and endocytosis.21 The exact molecular mechanism of the dynamins and Drp-1 in this process is still subject to debate. However, one of the models postulates that these proteins act as mechanoenzymes that actively participate in the sectioning of membranes by constriction.21
Drp-1 is a protein that is mainly distributed in the cytoplasm, but it has a fraction that is localized to specific points of the external mitochondrial membrane, points that will be future fission sites (Figure 2). Drp-1 lacks a mitochondrial target sequence, and so it is recruited to the membrane by Fis1. This adaptor protein participates in the assembly of fission complexes of high molecular weight.21 Currently, direct interaction between Fis1 and Drp-1 has only been demonstrated in recombinant proteins, unlike the case in yeasts, in which the mitochondrial localization of Dnm1p (Drp-1 homologue) is completely lost in Fis1p mutants (Fis1 homologue).22 Fis1p recruits Dnm1p to the mitochondrium through the molecular adaptors Mdv1p and Caf4p23; however, the presence of these proteins in mammals has not yet been demonstrated.
It is important to clarify that the mitochondrial fission process usually occurs in all cells in normal conditions. However, mitochondrial fission has also been associated with conditions of metabolic stress,24 as well as autophagy,25, 26 and apoptosis.27, 28
THE MACHINERY OF MITOCHONDRIAL FUSIONThe main regulators of mitochondrial fusion in humans are the mitofusin (Mfn) proteins and the optic atrophy-1 (OPA1) protein (Figure 2). Mfn has 2 isoforms (Mfn1 and Mfn2), which are localized in the external mitochondrial membrane with their N-terminal domain (GTPase domain) and C-terminal domain oriented towards the cytosol. Whereas Mfn1 and Mfn2 interact with each other to coordinate fusion of the external mitochondrial membrane of opposing mitochondria,29, 30 OPA1 is localized to the intermembrane space and associated with the internal mitochondrial membrane. OPA1 participates in the remodeling of the mitochondrial crests and the approach and fusion of the internal mitochondrial membrane.31, 32 The fusion of the 2 mitochondrial membranes seems to occur as 2 independent and separate events. Whereas fusion of the external mitochondrial membrane requires a low concentration of GTP, fusion of the internal membrane requires hydrolysis of GTP and an intact mitochondrial membrane potential (Ψmt) and, therefore, high ATP synthesis.33
Recent evidence shows that fusion directly regulates mitochondrial metabolism.34 Thus, a decrease in the concentration of OPA1 protein or of either Mfn by interference RNA leads to the formation of fragmented mitochondria with a lower oxygen uptake and a lower Ψmt.34 Although the function of these proteins (Mfns and OPA1) is known in mitochondrial fusion and remodeling, their relationship with the metabolic machinery is unknown. Likewise, it is not known why loss of some of these proteins directly interferes with cellular respiration. In contrast, overexpression of Mfn2 directly increases the activity of respiratory complexes, mitochondrial oxidation, and glucose utilization.35 Expression of Mfn2 is decreased in skeletal muscle of obese Zucker rats and in obese and diabetic patients. This highlights the pathological importance of these abnormalities in mitochondrial dynamics and morphology and provides evidence for the critical role of mitochondrial plasticity in maintaining the function of this organelle.36, 37
MITOCHONDRIAL DYNAMICS IN THE HEARTTissues with a high energy demand such as the heart and skeletal muscle tend to have a fused mitochondrial network, whereas those with a low energy demand, such as the liver, have a network where fission is more apparent.38 Mitochondrial dynamics in cardiac tissue is a little studied topic, perhaps because of the perception that it does not have an important role, given the highly structured nature of the cardiac cell.39
The adult cardiac myocyte has 2 groups of mitochondria according to its cytoplasmic location. The interfibrillary mitochondria (IFM) are interleaved in the contractile machinery (muscle fiber), whereas the subsarcolemmal mitochondria are located under the plasma membrane (sarcolemma).40 The existence of these populations implies that the mitochondria of the adult cardiac tissue do not form homogeneous networks as in the neonate cardiac myocyte. Interestingly, in the adult heart, there are more proteins implicated in mitochondrial dynamics than in the other tissues.41
Given the high oxidation index of cardiac myocytes, a possible relationship between mitochondrial dynamics, metabolism, and mechanical efficiency of the heart has been studied. Studies of mitochondria in adult cardiac myocytes have focused mainly on assessing the size and morphology of the organelle in pathophysiological conditions.42, 43 For example, the damage produced by ischemia-reperfusion is associated with a fissioned mitochondrial morphology and an increased opening probability of the mitochondrial transition pore.43 These effects are counteracted upon inhibiting the machinery of mitochondrial fission, and such an action decreases the extent of infarction in mice that undergo occlusion of the coronary artery.43
Similarly, in diabetic cardiomyopathy, there are functional abnormalities in the mitochondria, such as decreased respiration and lower expression of proteins involved in oxidative phosphorylation and production of ATP.42
In another study of diabetic cardiomyopathy, it was observed that the mitochondrial abnormalities occurred mainly in the IFM subpopulation.44 In this experimental model, the size, activity of mitochondrial complexes I and III, and the concentration of cardiolipin were lower, whereas superoxide production and lipid peroxidation were increased only in the IFM.44 Given that the IFM generate ATP to sustain contraction, the decreased metabolic activity of this subpopulation could be particularly harmful by contributing to loss of mechanical efficiency in hearts with diabetic cardiomyopathy.44
HEART FAILURE AND MITOCHONDRIADuring the development of heart failure, the heart undergoes extensive metabolic remodeling and its preference for energy substrates changes. Thus, as disease progresses, oxidation of FFAs decreases slowly45 which is associated with a progressive decrease in the quantity of ATP in the heart.2, 46 In the early stages of heart failure, the concentrations of ADP and AMP increase, and this promotes greater uptake and utilization of glucose through activation of AMP-activated protein kinase.47 This change temporarily reduces the metabolic demands of the cardiac myocytes; however, the quantity of ATP generated by glycolysis is lower than that produced by β-oxidation, and so the cardiac ATP content inevitably decreases during the terminal stages of the disease.47 In parallel, the accumulation of unmetabolized FFAs is associated with a decrease in up to 30% of the mechanical efficiency of the myocardium.48
The current therapeutic alternatives for heart failure seek to enhance glucose utilization in the heart by directly inhibiting oxidation of FFAs or their mitochondrial import by carnitine-palmitoyltransferase-1. These pharmacological alternatives can help compensate for the loss of ATP in the heart.46, 47 Although most drugs available have complex mechanisms of action and these have not been fully elucidated (Table 1), all block FFA oxidation and promote glucose utilization.46, 47 While the effect of these interventions in heart failure depends on the etiology or stage of the disease,47 several clinical studies indicate that partial inhibition of β-oxidation is a promising add-on alternative to neurohumoral blockage.57, 58
Table 1. Drugs That Modify Substrate Utilization in the Heart.
| Drug | Proposed mechanism | Effect | Reference |
| Etomoxir | Inhibition of CPT-1 | Improves EF and quality of life in HF. Requires more studies in humans. | Palaniswamy et al. 49 ; Schimdt et al. 50 |
| Oxfenicin | Inhibition of CPT-1 | Inhibits remodeling in animal models of HF. Produces dose-dependent hypertrophy by an unknown mechanism. | Greaves et al. 51 ; Lionetti et al. 52 |
| Perhexiline | Inhibition of CPT-1 and CPT-2 | Improves EF and quality of life in HF. Neuro- and hepatotoxic, requieres studies of long-term administration. | Palaniswamy et al. 49 ; Lee et al. 53 |
| Ranolazine | Partial inhibition of β-oxidation | Improves EF in animals | Palaniswamy et al. 49 ; McCormack et al. 54 |
| Trimetazidine | Inhibition of 3-CAT (CPT-1?) | Improves EF and quality of life in HF. Requires more studies in humans. | Palaniswamy et al. 49 ; Rosano et al. 55 ; Wenmeng et al. 56 |
3-CAT, 3-ketoacyl coenzyme A mitochondrial thiolase; CPT-1, carnitine palmitoyltransferase 1; CPT-2, carnitine palmitoyltransferase 2; EF, ejection fraction; HF, heart failure.
Given the high energy consumption of the cardiac myocyte, there is a close relationship between cardiac and mitochondrial function. The clinical evidence indicates that mitochondrial energy metabolism plays a critical role in different heart diseases (Table 2).
Table 2. Cardiac Diseases Caused by Mitochondrial Disorders.
| Primary defect | Locus affected | Cardiac phenotype |
| Oxidative phosphorylation | ||
| Point mutations in mtDNA | Several loci | DCM/HCM |
| Loss of mtDNA | Several loci | DCM/HCM |
| Assembly of COX enzymes | SCO2 | HCM |
| SKS/loss of mtDNA | Sporadic mutation | DCM |
| Oxidation of fatty acids | ||
| CPT-2 deficiency | CPT-2 | CM/arrhythmia |
| SCAD deficiency | SCAD | Childhood CM |
| MTP deficiency (includes LCHAD defects) | MTPa, MTPb subunits | Childhood CM/arrhythmia |
| VLCAD deficiency | VLCAD | Childhood CM |
| CPT-1 deficiency | L-CPT-1 | CM |
| Carnitine transport | OCTN2 | Childhood CM |
| Carnitine translocase deficiency | CAC | Childhood CM/arrhythmia |
| Others | ||
| Friedreich ataxia | Frataxin | HCM |
| Barth syndrome | G4.5 | DCM |
| Mutation in the mitochondrial phosphate transporter (Mayr et al. 60 ) | SLC25A3 | HCM |
| Light chain mutation of myosin (Poetter et al. 61 ) | MYL3 | HCM |
| Mitochondrial tRNA mutations | Leu | DCM/HCM |
| Ile | Childhood CM/HCM | |
| Lis | HCM |
CM, cardiomyopathy; COX, cytochrome C oxidase; CPT, carnitine palmitoyltransferase; DCM, dilated cardiomyopathy; HCM, hypertrophic cardiomyopathy; Ile, isoleucin; LCHAD, long chain 3-hydroxyacyl-coenzyme A dehydrogenase; Leu, leucine; Lis, lysine; mtDNA, mitochondrial DNA; MTP, mitochondrial trifunctional protein; SCAD, short-chain acyl coenzyme A dehydrogenase; SKS, Kearns-Sayre syndrome; tRNA, transfer RNA; VLCAD, very long chain acyl coenzyme A dehydrogenase.
Adapted from Marín-García et al. 59
Specific mutations in genes coding for mitochondrial proteins such as ANT (adenine nucleotide translocator, which exchanges mitochondrial ATP for cytosolic ADP), subunits of respiratory complexes, β-oxidation enzymes, and molecules associated with assembly of complex IV, among others, are detected in patients with familial forms of dilated cardiomyopathy.59
Similar results have been replicated in different transgenic models, thereby reaffirming the relationship between mitochondrial and cardiac function.62 Deletion of the murine gene-encoding ANT leads to oxidative phosphorylation defects and a phenotype characterized by progressive cardiac hypertrophy.62 In addition, deletion of the mitochondrial transcriptional factor Tfam, essential for biogenesis and organelle function, triggers lethal embryonic cardiomyopathy, whereas heterozygous mice develop severe dilated cardiomyopathy and the animals are viable only until 20 days of development.63
HEART FAILURE AND MITOCHONDRIAL DYNAMICSIn recent years, evidence has arisen that links changes in mitochondrial morphology with development of heart diseases. Small and disorganized mitochondria are observed both in dilated cardiomyopathies64 and myocardial hibernation.65 Similar findings have been reported in experimental models of ischemic myocardia in which the mitochondrial network is fragmented, with a decrease in the organelle area along with an increase in the number of mitochondria.66
Changes in the mitochondrial morphology of cardiac myocytes have been reported in a range of cardiomyopathies. Cardiac hypertrophy arising from aortic constriction in rats is accompanied by increases in mitochondrial size and mitochondrial DNA replication.67 Likewise, in canine models of chronic heart failure, the number of small mitochondria increases, accompanied by loss of mitochondrial matrix density.68 This morphology is similar to that described in muscle biopsies of patients with mitochondrial disorders and in models of dilated cardiomyopathy, which are characterized by the presence of fragmented mitochondria and a decrease in OPA1.69 These results illustrate an association between morphological mitochondrial disorders and changes in the cellular bioenergetic capacity.
On the other hand, in biopsies of patients with idiopathic dilated cardiomyopathy, the presence of giant mitochondria is observed with a lower density of mitochondrial matrix, associated with an increase in their number.70 Similar results have been observed in experimental models of hypoxia,71 indicating that the mitochondrial network in the adult cardiac myocyte is effectively dynamic and that remodeling of this network is part of the pathological process.
Although there is no integrated model that takes into account the participation of the mitochondrial network in cardiac physiology, investigators are beginning to study the link between mitochondrial dynamics and cardiovascular disease in apoptotic processes, autophagy, metabolic regulation, and ischemia-reperfusion.43 These are described below.
Mitochondrial Morphology and ApoptosisFragmentation of the mitochondrial network in response to apoptotic stimuli is a finding observed in many cell types, and so an association between activation of the mitochondrial fission machinery and this cell death process has been postulated.27, 72 Our group has investigated this relationship in primary cultures of cardiac myocytes of neonate rats, and we have shown directly the existence of molecular mitochondrial fission machinery in cardiac cells and its relationship with early events in apoptosis induced by ceramides.28 The loss of integrity of the mitochondrial membrane leads to release of cytochrome C, endonuclease G, apoptosis inducing factor, and Smac, which activate the intrinsic apoptotic pathway.73 This event is mediated by a complex process that involves Drp-1, Mfn2, and proapoptotic protein BAX.74 Overexpression of antiapoptotic proteins Bcl2 attenuates apoptosis induced by ischemia-reperfusion75 and decreases the probability of mitochondrial transition pore opening in the event of calcium overload, improving the contractile phenotype in cardiomyopathic mice.76 On the other hand, the decrease in the concentration of Mfn2 fusion protein is associated with an increase in mitochondrial fragmentation. This promotes release of cytochrome C and points to a protective role of Mfn2 against apoptotic death induced by ceramides in this model.28
Mitochondrial Morphology and AutophagyAutophagy is a cellular response to conditions of nutrient deprivation and hypoxia that allows remodeling of organelles and proteins for reuse of amino acids and fatty acids in essential cellular functions.77 In addition, autophagy plays a role in cellular “quality control” by permitting elimination of aged and dysfunctional organelles or damaged proteins.78 Loss of Ψmt seems to be the main signal for degradation of individual mitochondrial units (mitophagy).25, 79 Although the mitochondrial fission and fusion processes seem to be essential for mitophagy,80 it has yet to be established whether regulation of the mitochondrial dynamics is associated with changes in autophagy processes in the cardiac myocyte.26
Several studies have shown that inhibition of autophagy in the cardiac myocyte promotes its death by apoptosis in response to nutrient deprivation and in models of ischemia-reperfusion, and this can contribute to deterioration in the contractile function characteristic of heart failure.81, 82
Mitochondrial Morphology and MetabolismAs has been already mentioned, the proteins involved in mitochondrial dynamics regulate mitochondrial function. The decreased expression of Mfn2 is associated with a decrease in substrate oxidation,35 respiration, and mitochondrial potential, findings similar to those reported for models with OPA134 and Drp-183 gene silencing. These findings have been partially replicated in animal models and in samples of myocardium in individuals with heart failure, and a significant decrease in proteins regulating mitochondrial dynamics was observed.66 Overall, the available evidence indicates that any interruption in mitochondrial dynamics has a negative impact on the function of this organelle.43 However, the pathogenic role of changes in the abundance of these proteins and their possible relationship with metabolic changes in the hearts of patients with heart failure have yet to be elucidated.
Mitochondrial Morphology in Cardiac Ischemia-ReperfusionThe evidence discussed above indicates that the ischemia-reperfusion model corresponds to a condition in which changes in the mitochondrial morphology are particularly relevant. Ong et al.43 reported that the inhibition of mitochondrial fission protects the cardiac myocyte against ischemia-reperfusion injury in both in vitro and in vivo models.43 Treatment with mdivi-1, a pharmacological inhibitor of Drp-1, increases the proportion of adult cardiac myocytes with elongated mitochondria and protects them against simulated ischemia-reperfusion by inhibiting mitochondrial transition pore opening and decreasing the infarcted area.43 These findings are in agreement with the reduction in the concentration of OPA1 fusion protein reported in samples from hearts with ischemic cardiomyopathy.66 This is the first direct evidence that modulation of mitochondrial morphology could be a new therapeutic alternative in cardiac disease. However, the potential protective role of this intervention in heart failure has still not been assessed.
OUTLOOKIn recent years, the traditional view of mitochondria as discrete and independent units has changed substantially to give rise to a model of a mitochondrial network, which undergoes dynamic and active remodeling through fusion and fission processes in response to numerous pathophysiological stimuli. “Mitochondrial dynamics” is an integral part of the cellular metabolic flexibility. In fact, through studies of interventions in the machinery of mitochondrial dynamics, there is evidence of a strong association between morphology and function in this organelle. In the cardiovascular system, there is consensus that the processes of mitochondrial dynamics can participate in the development of cardiovascular disease. Alterations in the equilibrium between fusion and fission are a common characteristic of many cardiac diseases that end up in heart failure. Given this close relationship between mitochondrial dynamics and function, an emerging therapeutic target and one that is complementary to neurohumoral blockade is remodeling of cardiac metabolism. Initial studies have been based on pharmacological intervention in the metabolism of substrates, with promising results regardless of the etiology of heart failure. However, it has not been clarified whether part of the benefit of heart failure therapies, including metabolic interventions, is associated with changes in the mitochondrial network. On the other hand, development of specific interventions directed towards modulating mitochondrial dynamics could be clinically relevant and complementary. A basic challenge in current cardiology research is exhaustive investigation of the molecular mechanisms of this pathological condition in the search of new therapeutic alternatives.
FUNDINGJovan Kuzmicic, Andrea del Campo, Camila López-Crisosto, Pablo E. Morales, Christian Pennanen, Roberto Bravo-Sagua, Ramiro Zepeda, Hugo E. Verdejo and Valentina Parra received grants from CONICYT, Chile. These studies have been funded by the FONDECYT projects 1080436 (Sergio Lavandero), FONDAP 15010006 (Sergio Lavandero), FONDECYT 1090727 (Pablo F. Castro), and FONDECYT 1110180 (Mario Chiong).
CONFLICT OF INTERESTSNone declared.
Corresponding author: Centro Estudios Moleculares de la Célula, Facultad de Ciencias Químicas y Farmacéuticas, Universidad de Chile, Olivos 1007, 8380492 Santiago, Chile. slavander@uchile.cl