
Hypertrophic cardiomyopathy (HCM) is a disorder with variable expression. It is mainly caused by mutations in sarcomeric genes but the phenotype could be modulated by other factors. The aim of this study was to determine whether factors such as sex, systemic hypertension, or physical activity are modifiers of disease severity and to establish their role in age-related penetrance of HCM.
MethodsWe evaluated 272 individuals (mean age 49 ± 17 years, 57% males) from 72 families with causative mutations. The relationship between sex, hypertension, physical activity, and left ventricular hypertrophy was studied.
ResultsThe proportion of affected individuals increased with age. Men developed the disease 12.5 years earlier than women (adjusted median, 95%CI, –17.52 to –6.48; P < .001). Hypertensive patients were diagnosed with HCM later (10.8 years of delay) than normotensive patients (adjusted median, 95%CI, 6.28-17.09; P < .001). Individuals who performed physical activity were diagnosed with HCM significantly earlier (7.3 years, adjusted median, 95%CI, –14.49 to –1.51; P = .016). Sex, hypertension, and the degree of physical activity were not significantly associated with the severity of left ventricular hypertrophy. Adjusted survival both free from sudden death and from the combined event were not influenced by any of the exploratory variables.
ConclusionsMen and athletes who are carriers of sarcomeric mutations are diagnosed earlier than women and sedentary individuals. Hypertensive carriers of sarcomeric mutations have a delayed diagnosis. Sex, hypertension, and physical activity are not associated with disease severity in carriers of HCM causative mutations.
Keywords
Hypertrophic cardiomyopathy (HCM) is a genetic cardiac disease characterized by morphological, functional, clinical, and prognostic heterogeneity.1–7 This variable phenotypic expression and its incomplete penetrance8 have constituted an obstacle to a full understanding of the clinical spectrum and the consequences of the disease. HCM is mainly caused by mutations in genes encoding sarcomeric contractile proteins.9,10
Pathogenic mutations have been identified in more than 12 genes.11MYH7 and MYBPC3 gene mutations account for about 80% of genotyped cases.12,13
Although intense and competitive exercise is not recommended in the clinical guidelines for HCM,14 the association between physical activity and outcomes in HCM is weak. Nevertheless, physical activity has been accepted as a trigger for ventricular arrhythmias and sudden death (SD) in patients with HCM.15 Clinicians are confronted with HCM patients, or even unaffected carriers, seeking advice on physical activity. Discerning whether lifestyle choices can modify penetrance is crucial.16
Sex has proven to be an important variable in the natural history of the characterization and management of a variety of acquired cardiovascular conditions.17–22
Similar to intense physical activity, hypertension (HT) is thought to enhance the degree of left ventricular hypertrophy (LVH) via the induction of cardiomyocyte hypertrophy.23
Observations of within-family variations are not explained by mutational heterogeneity and therefore environmental factors must be involved.21
The primary aim of this study was to compare penetrance, understood as age at diagnosis of HCM, clinical characteristics, and outcomes in a large cohort of genotyped HCM families with mutations in MYBPC3 and MHY7, in terms of sex, the presence or absence of HT, and the intensity of physical activity.
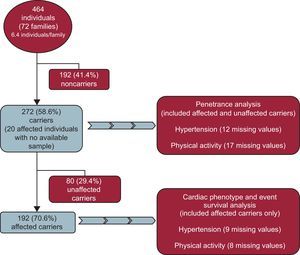
METHODSStudy SampleAll participants were recruited from an initial pool of 72 families (464 individuals) with HCM; From these 72 families, we excluded 192 (41.4%) normal relatives who did not carry the family mutation. In the sample of 272 carriers from 72 different families, 20 affected individuals with clinical variables but no available sample were included as obligate carriers (Figure 1).

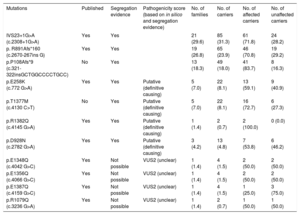
Our cohort comprised a total of 272 individuals (mean age 49.0 ± 17.5 years, 57% males). Two hundred and twenty-one (81.3%) were carriers of a causative mutation in the MYBPC3 gene and 51 (18.8%) in the MYH7 gene (Table 1).
Mutation Frequency in our MYH7-MYBPC3 Carriers
| Mutations | Published | Segregation evidence | Pathogenicity score (based on in silico and segregation evidence) | No. of families | No. of carriers | No. of affected carriers | No. of unaffected carriers |
|---|---|---|---|---|---|---|---|
| IVS23+1G>A (c.2308+1G>A) | Yes | Yes | 21 (29.6) | 85 (31.3) | 61 (71.8) | 24 (28.2) | |
| p. R891Afs*160 (c.2670-267ins G) | Yes | Yes | 19 (26.8) | 65 (23.9) | 46 (70.8) | 19 (29.2) | |
| p.P108Afs*9 (c.321-322insGCTGGCCCCTGCC) | No | Yes | 13 (18.3) | 49 (18.0) | 41 (83.7) | 8 (16.3) | |
| p.E258K (c.772 G>A) | Yes | Yes | Putative (definitive causing) | 5 (7.0) | 22 (8.1) | 13 (59.1) | 9 (40.9) |
| p.T1377M (c.4130 C>T) | No | Yes | Putative (definitive causing) | 5 (7.0) | 22 (8.1) | 16 (72.7) | 6 (27.3) |
| p.R1382Q (c.4145 G>A) | Yes | Yes | Putative (definitive causing) | 1 (1.4) | 2 (0.7) | 2 (100.0) | 0 (0.0) |
| p.D928N (c.2782 G>A) | Yes | Yes | Putative (definitive causing) | 3 (4.2) | 13 (4.8) | 7 (53.8) | 6 (46.2) |
| p.E1348Q (c.4042 G>C) | Yes | Not possible | VUS2 (unclear) | 1 (1.4) | 4 (1.5) | 2 (50.0) | 2 (50.0) |
| p.E1356Q (c.4066 G>C) | Yes | Not possible | VUS2 (unclear) | 1 (1.4) | 4 (1.5) | 2 (50.0) | 2 (50.0) |
| p.E1387Q (c.4159 G>C) | Yes | Not possible | VUS2 (unclear) | 1 (1.4) | 4 (1.5) | 1 (25.0) | 3 (75.0) |
| p.R1079Q (c.3236 G>A) | Yes | Not possible | VUS2 (unclear) | 1 (1.4) | 2 (0.7) | 1 (50.0) | 1 (50.0) |
VUS, variant of unknown clinical significance (VUS2, uncertain).
Values are expressed as No. (%).
A founder effect of c.2308+1G>A, p.P108Afs*9 and p.R891Afs*160 (c.2670-267insG) in MYBPC3 was demonstrated with haplotype analysis. Seventy-four percent of the carriers shared 1 of these 3 founder mutations from the region, which are thought to cause a truncated protein. Cosegregation and/or in silico studies in all variants were demonstrated and were consistent with a disease-causing mutation (Table 1). Eight individuals (2.9%) had an additional mutation in another gene.
Hypertrophic cardiomyopathy was defined in probands as unexplained LVH (≥ 15mm) in the absence of another cardiac or systemic disease able to cause LVH.1 Relatives were considered to be affected if they fulfilled the diagnostic criteria within the context of familial HCM.24,25
Patients were prospectively included and cardiac imaging and examinations were performed prospectively; symptoms and arrhythmic events prior to the first evaluation were also recorded. The mean follow-up was 5.5 ± 3.3 years.
After cardiac examination, 192 (70.6%, mean age 52.5 ± 16.6 years, 127 [66.1%] males) met the HCM diagnostic criteria and were considered clinically affected (patients), while 80 (29.4%) were considered normal (all carriers).
Of the 260 individuals evaluated, 63 (23.2%) had HT. Information on blood pressure was lacking in 12 (4.4%). There were 58 (31.7%) hypertensive and 125 (68.3%) normotensive individuals among the 183 HCM-affected patients with available information. Among the 77 normal carriers with available values, there were 5 (3.3%) hypertensive and 72 (90%) normotensive individuals.
Physical ActivityIndividuals were asked about their physical activity and were classified according to a “typical week” concept. Only the hours of physical activity estimated to be associated with an increase of > 70% or maximal heart rate were considered. Physical activity during the last 2 years prior to the time of the diagnosis in affected carriers or to the time of first evaluation in unaffected carriers was considered. Interviews were performed prospectively at the time of the first evaluation for patients seen from 2007 to 2015 and those patients evaluated from 2003 to 2006 were contacted by telephone. Patients who were sedentary at the time of evaluation but had a history of participating in competitive sports in the past were reclassified as physically active.
- •
Group 1 (n = 156 [84.9%]). Sedentary or low physical activity (generally without sweating) (eg, walking, gardening): included sedentary lifestyle participants (n = 137 [74.5%]) or individuals who exercised for ≤ 2hours of significant physical activity/wk (n = 19 [10.3%]).
- •
Group 2 (n = 17 [9.3%]). Intermediate or moderate physical activity: included individuals who performed significant physical activity for > 2 and < 5h/wk (n = 13 [7.1%]). Those who had worked in a physically demanding job (n = 4 [2.2%]) were also included in this group.
- •
Group 3 (n = 11 [6%]). Intense or high physical activity: included amateur athletes, professional (n = 6 [3.3%]) and ex-professional athletes (n = 5 [2.7%]) who performed intense regular physical activity for > 5h/wk.
Sixty (84.5%) out of the 71 unaffected carriers were sedentary or performed low physical activity, 8 (11.3%) performed intermediate or moderate physical activity, and 3 (4.2%) performed intense or high physical activity. In 9 (11.2%), no information on physical activity was available.
Table 1 of the supplementary material shows details of the number of patients for each of the subgroups regarding exposure variables (HT, sex, and physical activity), according to the classification of “affected carrier”/“unaffected carrier”. An expanded Methods section is available in the Methods section of the supplementary material.
Statistical AnalysisThe statistical analysis was performed using SPSS (version 15.0) statistical software and STATA (version 13) for Laplace survival analysis.
Age at diagnosis, maximal ventricular hypertrophy, and SD were defined as dependent variables in the analysis. Sex, HT, and physical activity were defined as exploratory variables on multivariate analysis. Sudden death equivalent was defined as SD, resuscitated cardiac arrest, or the presence of an implantable cardioverter-defibrillator. New York Heart Association (NYHA) class III-IV includes advanced heart failure symptoms, heart transplant, and heart failure-related death.
Multivariate survival analysis included all 3 exploratory variables and those selected as covariates. For penetrance (age at HCM diagnosis), the “type of gene” (MYBPC3 vs MYH7) was included in the model as an additional covariate. For stroke, atrial fibrillation (AF), “heart failure (NYHA III-IV)”, “SD equivalent” and “combined event” survival analysis, a list of covariates included indexed maximal wall thickness (mm/m2), left atrial diameter (mm), the presence of left ventricular outflow tract obstruction (> 30mmHg), and type of gene. Laplace derived analysis26 was used for survival analysis and Figure 2 and Figure 3. The median was given as a summary of the comparisons. Linear logistic regression was used for the evaluation of the impact of the exploratory variables on LVH (maximal wall thickness in mm). P-values < .05 were considered statistically significant.
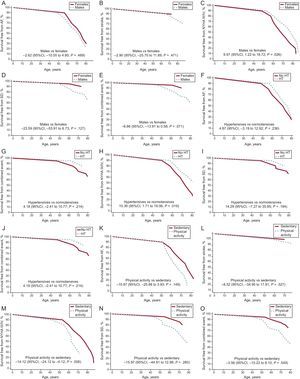
 in carriers of MYBPC3 and MYH7 mutations. A: penetrance by sex; B: presence or absence of HT; C: intensity of physical activity. Adjusted Laplace median and 95%CI are given. All 3 exploratory variables (sex, HT, and physical activity) and “type of gene” were included in the analysis of each of the graphs. All genetic carriers, affected and unaffected, were included in the analysis. 95%CI, 95% confidence interval; HCM, hypertrophic cardiomyopathy; HT, hypertension.")
Penetrance of the disease (HCM) in carriers of MYBPC3 and MYH7 mutations. A: penetrance by sex; B: presence or absence of HT; C: intensity of physical activity. Adjusted Laplace median and 95%CI are given. All 3 exploratory variables (sex, HT, and physical activity) and “type of gene” were included in the analysis of each of the graphs. All genetic carriers, affected and unaffected, were included in the analysis. 95%CI, 95% confidence interval; HCM, hypertrophic cardiomyopathy; HT, hypertension.
![Adjusted Laplace estimates of survival. A: free from AF; B: stroke; C: NYHA III-IV; D: SD; E: the combined event by sex; F: free from AF; G: stroke; H: NYHA III-IV; I: SD; J: the combined event for the presence or absence of HT; K: free from AF; L: stroke; M: NYHA III-IV; N: SD; O: the combined event by physical activity. Laplace median adjusted value and 95CI are given. All 3 exploratory variables (sex, HT, and physical activity) and covariates (indexed maximal wall thickness [mm/m2], left atrial diameter [mm], presence of left ventricular outflow tract obstruction [> 30mmHg] and type of gene) were included for analysis of each of the graphs and analyses. Only affected carriers were included in the analysis. 95%CI, 95% confidence interval; AF, atrial fibrillation; HT, hypertension; NYHA, New York Heart Association; SD, sudden death.](https://static.elsevier.es/multimedia/18855857/0000007100000003/v1_201803151232/S1885585717303067/v1_201803151232/en/main.assets/gr3.jpeg?xkr=eyJpdiI6IjdPaTk3TE1ZdmNYK3hZUk14bTlpT2c9PSIsInZhbHVlIjoiM1NQc3N5US8yaDNIc1BUNFdnc2QrUGZRSkJySXYycEM0RlZ6ZDQ3K2NoNlJGUWxyRHVhbm1jbXpkNS84Q3ZNajZRWUUrSEQyYmxzTHhLaGl2Q0FDMzZDcFVLdlJKUnJta1FCU2tjKzFVMHFXRmpmb2VNOVA5Wm5HdVljdUxUZll0VXNlREpUZFVFTE5TeXd2NkhHYXFMRWYyQXA4WHkvTS9mNTZPcW5URStxV3oyaFNIeHVsNzVvUWhtbzcyTCtmTWxrWFBtbkVtaW1sNWdwRTh1bnpwVTRFQXNUaFhDVE12WkFWUWhER2cwQk1oUDdDWUt0MlBXcnB4WERpaklOM1crNTNmeHdVSGY2alA1WGltOWxKcS82L1VLV2FLdXdQM2hMdHMxbnk1TFU9IiwibWFjIjoiZWQyNmIxNmYwZGNlMjhhMTQxY2RkYWUxYTg4NjRjMmFmMmEzMDZhYTc4NDA0YTA2OTgzMDNkZmNkMzkwYWEzMyIsInRhZyI6IiJ9 "Adjusted Laplace estimates of survival. A: free from AF; B: stroke; C: NYHA III-IV; D: SD; E: the combined event by sex; F: free from AF; G: stroke; H: NYHA III-IV; I: SD; J: the combined event for the presence or absence of HT; K: free from AF; L: stroke; M: NYHA III-IV; N: SD; O: the combined event by physical activity. Laplace median adjusted value and 95CI are given. All 3 exploratory variables (sex, HT, and physical activity) and covariates (indexed maximal wall thickness [mm/m2], left atrial diameter [mm], presence of left ventricular outflow tract obstruction [> 30mmHg] and type of gene) were included for analysis of each of the graphs and analyses. Only affected carriers were included in the analysis. 95%CI, 95% confidence interval; AF, atrial fibrillation; HT, hypertension; NYHA, New York Heart Association; SD, sudden death.")
Adjusted Laplace estimates of survival. A: free from AF; B: stroke; C: NYHA III-IV; D: SD; E: the combined event by sex; F: free from AF; G: stroke; H: NYHA III-IV; I: SD; J: the combined event for the presence or absence of HT; K: free from AF; L: stroke; M: NYHA III-IV; N: SD; O: the combined event by physical activity. Laplace median adjusted value and 95CI are given. All 3 exploratory variables (sex, HT, and physical activity) and covariates (indexed maximal wall thickness [mm/m2], left atrial diameter [mm], presence of left ventricular outflow tract obstruction [> 30mmHg] and type of gene) were included for analysis of each of the graphs and analyses. Only affected carriers were included in the analysis. 95%CI, 95% confidence interval; AF, atrial fibrillation; HT, hypertension; NYHA, New York Heart Association; SD, sudden death.
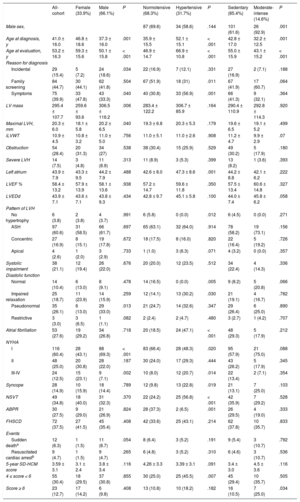
The clinical characteristics of the 192 patients at the first evaluation in terms of the 3 grouping variables (sex, HT, and physical activity) are summarized in Table 2.27
Main Clinical Features of the 192 Hypertrophic Cardiomyopathy Patients Evaluated
| All-cohort | Female (33.9%) | Male (66.1%) | P | Normotensive (68.3%) | Hypertensive (31.7%) | P | Sedentary (85.4%) | Moderate-intense (14.6%) | P | |
|---|---|---|---|---|---|---|---|---|---|---|
| Male sex, | 87 (69.6) | 34 (58.6) | .144 | 101 (61.6) | 26 (92.9) | .001 | ||||
| Age at diagnosis, y | 41.0 ± 16.0 | 46.8 ± 18.6 | 37.3 ± 16.0 | .001 | 35.9 ± 15.5 | 52.1 ± 15.1 | < .001 | 42.8 ± 17.0 | 32.2 ± 12.5 | .001 |
| Age at evaluation, y | 53.2 ± 16.3 | 59.3 ± 15.6 | 50.1 ± 15.8 | < .001 | 46.9 ± 14.7 | 66.9 ± 10.8 | < .001 | 55.0 ± 15.9 | 43.1 ± 15.2 | < .001 |
| Reason for diagnosis | ||||||||||
| Incidental | 29 (15.4) | 5 (7.2) | 24 (18.6) | .034 | 22 (16.9) | 7 (12.1) | .331 | 27 (16.9) | 2 (7.1) | .188 |
| Family screening | 84 (44.7) | 30 (44.1) | 62 (41.8) | .504 | 67 (51.9) | 18 (31) | .011 | 67 (41.9) | 17 (60.7) | .064 |
| Symptoms | 75 (39.9) | 33 (47.8) | 43 (33.3) | .040 | 40 (30.8) | 33 (56.9) | .001 | 66 (41.3) | 9 (32.1) | .364 |
| LV mass | 295.4 ± 107.7 | 259.6 ± 93.6 | 306.5 ± 116.2 | .006 | 283.4 ± 122.2 | 306.7 ± 85.9 | .164 | 290.4 ± 110.9 | 292.8 ± 114.3 | .920 |
| Maximal LVH, mm | 20.3 ± 6.0 | 18.1 ± 5.8 | 20.2 ± 6.5 | .040 | 19.3 ± 6.8 | 20.3 ± 5.3 | .179 | 19.6 ± 6.5 | 19.1 ± 5.2 | .499 |
| iLVWT | 10.9 ± 4.5 | 10.8 ± 3.2 | 11.0 ± 5.0 | .756 | 11.0 ± 5.1 | 11.0 ± 2.6 | .908 | 11.2 ± 4.7 | 9.9 ± 2.9 | .07 |
| Obstruction | 54 (28.4) | 20 (31.3) | 34 (27) | .538 | 38 (30.4) | 15 (25.9) | .529 | 49 (30.2) | 5 (17.9) | .180 |
| Severe LVH | 14 (7.5) | 3 (4.8) | 11 (8.9) | .313 | 11 (8.9) | 3 (5.3) | .399 | 13 (8.2) | 1 (3.6) | .393 |
| Left atrium | 43.9 ± 7.9 | 43.3 ± 9.5 | 44.2 ± 7.9 | .488 | 42.6 ± 8.0 | 47.3 ± 8.6 | .001 | 44.2 ± 8.8 | 42.1 ± 6.2 | .222 |
| LVEF % | 58.4 ± 13.2 | 57.9 ± 13.9 | 58.1 ± 13.6 | .938 | 57.2 ± 14.7 | 59.6 ± 11.8 | .350 | 57.5 ± 13.4 | 60.6 ± 14.8 | .327 |
| LVEDd | 43.9 ± 7.1 | 43.6 ± 7.1 | 43.8 ± 9.3 | .434 | 42.8 ± 9.7 | 45.1 ± 5.8 | .100 | 44.0 ± 7.4 | 45.8 ± 6.2 | .058 |
| Pattern of LVH | ||||||||||
| No hypertrophy | 6 (3.8) | 2 (3.8) | 4 (3.7) | .991 | 6 (5.8) | 0 (0.0) | .012 | 6 (4.5) | 0 (0.0) | .271 |
| ASH | 97 (60.6) | 31 (58.5) | 66 (61.7) | .697 | 65 (63.1) | 32 (64.0) | .914 | 78 (58.2) | 19 (73.1) | .156 |
| Concentric | 27 (16.9) | 8 (15.1) | 19 (17.8) | .672 | 18 (17.5) | 8 (16.0) | .820 | 22 (16.4) | 5 (19.2) | .726 |
| Apical | 4 (2.6) | 1 (2.0) | 3 (2.9) | .733 | 1 (1.0) | 3 (6.3) | .071 | 4 (3.2) | 0 (0.0) | .357 |
| Systolic impairment | 38 (21.1) | 12 (19.4) | 26 (22.0) | .676 | 20 (20.0) | 12 (23.5) | .512 | 34 (22.4) | 4 (14.3) | .336 |
| Diastolic function | ||||||||||
| Normal | 14 (10.4) | 6 (13.0) | 8 (9.1) | .478 | 14 (16.5) | 0 (0.0) | .005 | 9 (8.2) | 5 (20.8) | .066 |
| Impaired relaxation | 25 (18.7) | 11 (23.9) | 14 (15.9) | .259 | 12 (14.1) | 13 (30.2) | .030 | 21 (19.1) | 4 (16.7) | .782 |
| Pseudonormal | 35 (26.1) | 6 (13.0) | 29 (33.0) | .013 | 21 (24.7) | 14 (32.6) | .347 | 29 (26.4) | 6 (25.0) | .890 |
| Restrictive | 3 (3.0) | 3 (6.5) | 1 (1.1) | .082 | 2 (2.4) | 2 (4.7) | .480 | 3 (2.7) | 1 (4.2) | .707 |
| Atrial fibrillation | 53 (27.6) | 19 (29.2) | 34 (26.8) | .718 | 20 (18.5) | 24 (47.1) | < .001 | 48 (29.3) | 5 (17.9) | .212 |
| NYHA | ||||||||||
| I | 116 (60.4) | 28 (43.1) | 88 (69.3) | < .001 | 83 (66.4) | 28 (48.3) | .020 | 95 (57.9) | 21 (75.0) | .088 |
| II | 48 (25.0) | 20 (30.8) | 28 (22.0) | .187 | 30 (24.0) | 17 (29.3) | .444 | 43 (26.2) | 5 (17.9) | .345 |
| III-IV | 24 (12.5) | 15 (23.1) | 9 (7.1) | .002 | 10 (8.0) | 12 (20.7) | .014 | 22 (13.4) | 2 (7.1) | .354 |
| Syncope | 28 (14.9) | 10 (15.9) | 18 (14.4) | .789 | 12 (9.8) | 13 (22.8) | .019 | 21 (13.1) | 7 (25.0) | .103 |
| NSVT | 49 (34.8) | 18 (40.0) | 31 (32.3) | .370 | 22 (24.2) | 25 (56.8) | < .001 | 42 (35.9) | 7 (29.2) | .528 |
| ABPR | 30 (27.5) | 9 (29.0) | 21 (26.9) | .824 | 28 (37.3) | 2 (6.5) | .001 | 26 (29.5) | 4 (19.0) | .333 |
| FHSCD | 72 (37.5) | 27 (41.5) | 45 (35.4) | .408 | 42 (33.6) | 25 (43.1) | .214 | 62 (37.8) | 10 (35.7) | .833 |
| Events | ||||||||||
| Sudden deatha | 12 (6.3) | 1 (1.5) | 11 (8.7) | .054 | 8 (6.4) | 3 (5.2) | .191 | 9 (5.4) | 3 (10.7) | .792 |
| Resuscitated cardiac arrestb | 9 (4.7) | 1 (1.5) | 9 (4.7) | .265 | 6 (4.8) | 3 (5.2) | .310 | 6 (4.6) | 3 (10.7) | .536 |
| 5-year SD-HCM score | 3.59 ± 3.1 | 3.1 ± 2.4 | 3.8 ± 3.4 | .116 | 4.26 ± 3.3 | 3.39 ± 3.1 | .091 | 3.4 ± 3.0 | 4.5 ± 3.6 | .116 |
| 4 ≤ score < 6 | 55 (30.4) | 18 (29.5) | 37 (30.8) | .855 | 30 (25.0) | 25 (45.5) | .007 | 45 (29.4) | 10 (35.7) | .505 |
| Score ≥ 6 | 23 (12.7) | 17 (14.2) | 6 (9.8) | .408 | 13 (10.8) | 10 (18.2) | .182 | 16 (10.5) | 7 (25.0) | .034 |
ABPR, abnormal blood pressure response; ASH, asymmetrical septal hypertrophy; FHSCD, family history of sudden cardiac death; ICD, implantable cardioverter-defibrillator; iLVWT, indexed left ventricular wall thickness; LV, left ventricular; LVEDd, left ventricular end diastolic diameter; LVEF, left ventricular ejection fraction, LVH, left ventricular hypertrophy; NSVT, nonsustained ventricular tachycardia; NYHA, New York Heart Association; SD-HCM, sudden death-hypertrophic cardiomyopathy.
LV mass, by Deveraux formula (g); Obstruction, if left ventricular outflow tract gradient (> 30mmHg); Pattern of LVH, morphological subtype of hypertrophy according to Binder et al.27; Severe LVH, if Max LVH ≥ 30mm.
Values are expressed as mean ± standard deviation or No. (%).
Women had higher NYHA functional class at baseline and during follow-up. Fifteen (23.1%) women vs 9 (7.1%) men were in NYHA functional class III-IV (P = .002). IMLVWT, the percentage of obstruction, systolic and diastolic function variables, and the proportion of nonsustained ventricular tachycardia on Holter monitoring were all similar in women and men (Table 2).
The proportion of affected individuals increased with age. Men were diagnosed with the disease 12.50 years earlier than women (Laplace median 95%CI, –17.52 to –6.48; P < .001), adjusted for HT, physical activity, and “type of gene” (Figure 2A).
There were no differences in survival free from AF, stroke, NYHA functional class III-IV, SD, and the combined event (AF, stroke, NYHA III-IV, SD) in terms of sex (Figures 3A-3E).
HypertensionOf the 183 participants with available HT information and who met the HCM criteria, 58 (31.7%) had HT (Table 2). The presence of symptoms was the main reason for HCM diagnosis in hypertensive vs normotensive patients (33 patients [56.9%] vs 40 [30.8%]; P = .001). HT-HCM patients had worse NYHA class (12 hypertensive patients [20.7%] vs 10 normotensive patients [8.0%] had NYHA III-IV; P = .01) and reported more syncopes (13 [22.8%] vs 12 [9.8%]; P = .014). There was no difference in LVH (LV mass, MLVWT or IMLVWT) or left ventricular outflow tract obstruction. HT-HCM patients had apical LVH slightly more frequently. Left atrium and left ventricular end diastolic diameters were significantly enlarged in patients with HT-HCM compared with normotensive patients (P = .001 and P = .082, respectively). Patients with HT had more atrial and ventricular arrhythmias. Twenty-four (47.1%) HT-HCM patients had AF vs 20 (18.5%) in the normotensive group (P < .001). Nonsustained ventricular tachycardia on Holter monitoring was also more frequent among HT-HCM patients (25 patients [56.8%] vs 22 [24.2%]; P < .001). However, abnormal blood pressure response was more prevalent among normotensive patients (28 patients [37.3%] vs 2 [6.5%]; P =.001).
Diagnosis of HCM was delayed in hypertensive patients. Hypertensive patients had a later diagnosis of HCM, by 10.83 years, compared with normotensive patients (Laplace median 95%CI, 6.28-17.09; P < .001), adjusted by sex, physical activity, and “type of gene” (Figure 2B).
There was no difference between HT-HCM and normotensive patients in terms of survival free from AF or stroke (Figure 3F and Figure3G). Paradoxically, survival free from NYHA III-IV was lower in normotensive vs HT-HCM patients (P = .02) (Figure 3H), with no significant differences in SD or the combined event (AF, stroke, NYHA III-IV, SD) (Figure 3I and Figure3J).
Physical ActivityHypertrophic cardiomyopathy patients who performed intense physical activity were significantly younger at evaluation than more sedentary groups (43.1 ± 15.2 vs 55.0 ± 15.9 years; P = .001). There was no association between exercise intensity and LVH or left ventricular outflow tract obstruction. Of note, LA dimension and left ventricular end diastolic diameter were similar in both groups. There was a trend toward better diastolic function in the group of HCM patients who performed more intense physical activity (normal mitral flow 5 [20.8%] vs 9 [8.2%]; P = .066).
There was no significant difference in symptoms regarding the intensity of physical activity. The proportion of patients with nonsustained ventricular tachycardia on Holter monitoring and AF was similar in both groups.
Although the proportion of affected individuals was similar regardless of the intensity of physical activity (72.2% vs 71.8%), patients who performed more intense physical activity were diagnosed with HCM 7.34 years earlier than those in the sedentary group (Laplace median 95%CI, –14.49 to –1.51; P = .016), adjusted for sex, HT, and “type of gene” (Figure 2C).
There was no difference between these 2 groups in terms of survival free from AF, stroke, NYHA III-IV, SD, or the combined event (AF, stroke, NYHA III-IV, SD) (Figure 3K and Figure3O).
Table 2 of the supplementary material shows the clinical characteristics of 14 individuals who performed intense or competitive exercise (11 affected individuals + 3 unaffected carriers).
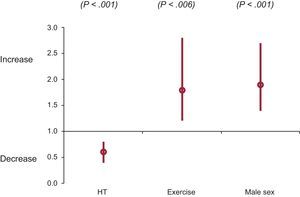
Multivariate AnalysisIn summary, sex, HT or physical activity were not predictors of LVH in the univariate or multivariate analysis. All 3 variables were independently associated with age at HCM diagnosis in Cox regression and in Laplace survival analysis (Figure 4). While male sex and intense physical activity were predictors of an earlier diagnosis, HT was associated with a delayed diagnosis.
 including sex, HT, and physical activity. 95%CI, 95% confidence interval; HT, hypertension.")
None of the 3 exploratory variables (sex, HT, physical activity) were associated with the events evaluated (stroke, AF, NYHA III-IV, SD, or combined event) in Cox regression analysis when the “type of gene”, left atrial diameter, indexed LVH and obstruction were included as covariables (Table 3 of the supplementary material). Left atrial diameter (mm) was the only covariate consistently associated with the events evaluated. Left ventricular hypertrophy together with left atrial diameter were independent predictors of SD in the Cox regression analysis (HR, 1.10; 95%CI, 1.00-1.20; P = .039; HR, 1.06; 95%CI, 1.03-1.10; P < .001), for indexed LVH and left atrial diameter, respectively).
When Laplace multivariate analysis was performed, left atrial diameter remained the main covariate associated with survival free from stroke, AF, NYHA III-IV, and the combined event but not with SD (Table 2 of the supplementary material and Figure 3). Laplace analysis also showed a value for the 3 exploratory variables on survival free from NYHA III-IV. None of the exploratory variables or covariates was associated with survival free from the combined event with this test.
DISCUSSIONEpigenetic mechanisms and environmental factors are fundamental in cardiac adaptations and disease.28
This study presents data on disease expression, penetrance–understood as the age at which HCM is diagnosed–and outcome in a large cohort of carriers of HCM-associated mutations. This is the first and largest study addressing the issue of HT and sex in terms of penetrance and severity of the phenotype and is the only study to address the impact of intense physical activity at the time of diagnosis, disease severity, and outcomes in a genotyped HCM series. Our study included 272 genotyped patients with 3 founder mutations with a very similar impact on the protein (MYBPC3 IVS23+1G>A, p.R891Afs*160, p.P108Afs*9), accounting for 74% of the population (n = 199, 85 + 65 + 49).
SexIn keeping with previous studies,17–21,29–32 we found that male sex was associated with an earlier diagnosis of HCM. In our series, men were diagnosed more than a decade earlier than women. Possible explanations for this consistent finding might be found in the role of estrogens in the development of hypertrophy.33
Once the phenotype developed, women behaved similarly to men, achieving similar degrees of LVH (indexed) and obstruction, and were even more limited symptomatically. We failed to identify consistent differences in heart failure, AF, and stroke survivals between the sexes. Consistent with previous literature, no significant differences were evidenced in SD and the combined event between the sexes in our series.
HypertensionThere is a general belief, based on indirect data or from small series, that HT should be associated with an increase in LVH in HCM patients34–36 and that intense physical activity is associated with an increase in LVH and is also a trigger for malignant arrhythmias and hence, a cause of SD in HCM.37,38
A recently published article on the role of HT and sex in penetrance and phenotype demonstrated a bivariate association between HT and HCM expression.35 The study failed to demonstrate differences between men and women and physical activity. Multivariate analysis was not possible due to the small sample size and there was a significant proportion of missing data (44%). In another recent article,36 no statistical analysis was performed. Physical activity was not recorded in this second series either.
To the best of our knowledge, this is the first study to clearly demonstrate the effect of additional risk factors on phenotypic outcome in a homogeneous HCM genotyped population. Hypertensive individuals in our series had a delayed diagnosis of HCM. This finding was not entirely unexpected, as hypertrophy could be considered a consequence of HT in some hypertensive patients with mild phenotypes. Moreover, medication given to HT patients could prevent or delay the occurrence of the HCM phenotype. The design of the study precludes further conclusions. While age at diagnosis was available for the analysis, age at the development of LVH was not.
Although HCM patients with HT were more symptomatic and had a higher prevalence of arrhythmias, that did not seem to have an impact on prognosis in the survival analysis compared with normotensive patients after adjustment with covariates.
Physical ActivityDiagnosis of HCM is included in the current guidelines as a cause of disqualification in competitive sports.39–41 This recommendation, which extends to silent carriers, is based on the hypothesis that regular exercise training and competitive sports can play a role in triggering cellular mechanisms leading to the HCM phenotype and arrhythmic outcomes in the presence of a predisposing gene abnormality. The authors of these guidelines conclude that there is no firm data to support this recommendation and that it is based on what seems to be most reasonable to expert panels.42
In our series, we found that carriers of sarcomeric mutations from the more intense exercise groups were diagnosed with HCM 7 years earlier than sedentary carriers.
Once the disease develops, HCM patients who perform physical activity in the higher intensity groups develop a similar degree of hypertrophy. Of interest, in the multivariate analysis, sex, HT and physical activity intensity were not predictors of LVH in our series. Interestingly, we failed to identify differences in the left ventricle and left atrium dimension between the exercise and sedentary HCM groups. Hypertrophic cardiomyopathy might prevent heart remodeling in the hearts of athletes. Physical activity seems to have more of an impact on right ventricle size in normal individuals and in desmosomal mutation carriers than it does on the left chamber in HCM patients.42 In contrast, all 3 variables were significantly associated with age at HCM diagnosis.
There is little information in the literature on the assumed poor prognosis of HCM patients who engage in intense physical activity and this comes from clinical reports and retrospective series.43
In our study, HCM patients who performed more intense physical activity had a similar rate of events (AF, stroke, NYHA III-IV, SD, and the combined event).
In conclusion, on the basis of our data, physical activity does play a role in age at diagnosis but is not significant in disease expression and, in particular, does not seem to have a major impact on prognosis. An explanation for this finding should be sought in future larger prospective studies.
LimitationsThe effect of double mutations could not be assessed, because most genetic studies were performed in the preultrasequencing era. Analysis of 2 to 5 genes (all including MYH7 and MYBPC3) was performed in 80% of the patients, while in 20% of the index cases, the genetic study was considered complete when 1 of the founder mutations was identified. Age at disease development could not be assessed because the study period was limited. Age at diagnosis was used as a surrogate of disease penetrance. Definitions for the intensity of physical activity were arbitrary. The classification aimed to evaluate the impact of physical habits at the time of diagnosis and disease severity on outcomes. Analysis of the dosage-effect of sport or physical activity was beyond the scope of this article. The number of individuals in the intense physical activity group was small.
CONCLUSIONSMen and carriers of HCM mutations who perform intense physical activity are diagnosed significantly earlier than women and sedentary relatives. In contrast, hypertensive carriers of HCM mutations have a delayed diagnosis. Sex, HT and the degree of physical activity are not significantly associated with the severity of LVH in carriers of HCM causative mutations.
- –
Male carriers of HCM mutations develop the disease earlier in life than female carriers. There is a general belief that HT and intense exercise are associated with an increase in LVH in HCM patients. Indirect data support the idea that intense physical activity might trigger ventricular arrhythmias leading to SD in HCM patients.
- –
This study demonstrates that, in addition to sex, HT and physical activity also have an impact on age at diagnosis of HCM in genetic carriers. The role of HT and physical activity in the severity of phenotype and outcome is not clinically significant.
This study was funded by a grant from the Fundación Española del Corazon-Coca-Cola LTD (2007) and supported by a grant from the Fundación para la Formación e Investigación Sanitaria de la Región de Murcia. The investigators are part of a cardiovascular research network of the Carlos III Health Institute (RIC; RD12/0042/0049) and IMIB (Instituto Médico de Investigación Biosanitaria). M. Sabater was supported by a grant from ISCIII (RIC). The investigators are part of the clinical group of CIBERER and the University of Murcia.
CONFLICTS OF INTERESTNone declared.
.
The authors would like to thank Iván Gómez, M. José Oliva, María López, M. Carmen Olmo, M. José Antolinos and Miguel Pagan for their help in both the clinical and molecular work of this project. We particularly acknowledge the patients and the BioBank (PT13/0010/0018) from the Spanish National Biobanks Network (B.000859).