Keywords
INTRODUCTION
Heart failure (HF) is the most expensive cardiovascular pathology in industrialized countries and the most common cause of hospitalization in adults over 60 years of age. Despite therapeutic advances, the risk of death per year is 20%-50% depending on the population group. There is thus an evident need for new strategies which can alter the evolution of the condition, alleviate the symptoms and prolong the patient life. Apoptosis is one of the principal causes of death of cardiocytes and of the progression of HF.1 The possibility of reducing the loss of cardiocytes by inhibiting apoptosis is an important area of investigation to improve the current treatment of HF cases.
A number of studies have demonstrated that the depletion of anabolic hormones is frequent in men with HF, and that deficiencies of type 1 insulin growth factor (IGF-1) and testosterone are associated with a worse prognosis and greater mortality.2,4 Studies with cell cultures of H9c2 cardiocytes have shown that IGF-1, by means of a rapid, non-genomic mechanism, is capable of protecting cardiocytes from apoptosis induced by hyperosmotic stress.5 In skeletal muscle cells and neurons, testosterone also protects from cell death by a mechanism independent of androgen receptors,6,7 that is, independent of the classical genomic route established for testosterone. We currently know that testosterone stimulates cardiac contractility, induces direct vasodilatation and improves the peripheral endothelial function8; however, there are no published studies which treat the role of testosterone as a modulator of apoptosis in cardiomyocytes.
Spironolactone and eplerenone are 2 drugs which have been shown to improve the prognosis in patients with severe HF.9,10 Both drugs are aldosterone antagonists, but have notable structural differences: spironolactone is an antimineralocorticoid which is associated with progestogenic and anti-androgenic effects, while eplerenone is a derivative of spironolactone in which the progestogenic and anti-androgenic effects have been minimized, thus potentiating its selective binding to the aldosterone receptor.11,12 These differences in selectivity may produce a different behaviour in the possible protective role of testosterone described above.
The objective of this study was to investigate whether testosterone protects cardiocytes from apoptosis and its possible mechanisms, and whether spironolactone and eplerenone act have differently behaviour with respect to this beneficial effect of testosterone.
METHOD
Design
We studied the effects of testosterone and its co-treatment with spironolactone or eplerenone on the apoptosis induced by the addition of sorbitol in cell cultures of rat embryo cardiocytes of the H9c2 line. Sorbitol induces hyperosmotic stress, which is one of the principal mechanisms of tissue damage in pathological states such as ischaemia, septic shock and acidosis. We evaluated the protective effect of testosterone on induced apoptosis and later the effect of the addition of aldosterone receptors blockers, eplerenone and spironolactone. Apoptosis was determined by analysis of cellular viability, DNA fragmentation and activation of caspase-3, -8, and -9. We also administrated flutamide, an androgen receptor antagonist, in order to study the involvement of the androgen receptors, and we analysed the participation of the signalling routes SAPK/JNK, ERK 1/2 and p-38 mitogen-activated protein kinase (MAPK) to study the possible involvement of non-genomic mechanisms.
Reagents
Sorbitol, testosterone, flutamide, and other reagents were obtained from Sigma-Aldrich (St. Louis, MO, USA), unless otherwise specified. Eplerenone and spironolactone were supplied by Pfizer Inc. (New York, NY, USA). Cell culture reagents such as culture medium (DMEM), L-glutamine, foetal bovine serum, penicillin, streptomycin and trypsin/EDTA were obtained from Gibco (Invitrogen, Carlsbad, CA, USA). The a-actin and anti-IgG antibodies were supplied by Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibodies against the phosphorylated forms and total of SAPK/JNK, p38 and ERK1/2, as well as caspase-3, -8, and -9 were supplied by Cell Signaling Technology, Inc. (Danvers, MA, USA). The reagents for ECL detection, autoradiographic film and the molecular weight markers were obtained from Amersham Pharmacia Biotech (Piscataway, NJ, USA).
Cell Culture and Treatments
Cell line H9c2, obtained from rat embryo hearts, was generously provided by Dr. F. Fernández Belda (Biochemistry Department, University of Murcia, Spain). H9c2 cells were maintained in culture using standard protocols, at 37º C in a humid atmosphere with 95% air and 5% CO2, and in DMEM culture medium supplemented with 10% foetal bovine serum, 2 mM L-glutamine, 100 U/mL penicillin and 100 µg/ mL streptomycin. All treatments were performed in the absence of foetal bovine serum and antibiotics. Apoptosis was induced by hyperosmotic stress with sorbitol (3 h, 0.6 M, S). Testosterone (100 nM, T) was administered simultaneously with sorbitol unless otherwise indicated. Flutamide (10 µM, F), spironolactone (10 µM, Sp) and eplerenone (10 µM, E) were administered 30 min before the treatment with sorbitol and testosterone. The dose of flutamide chosen has been previously validated by other investigators,13 while the doses of spironolactone and eplerenone were validated by means of testing with concentrations in the range 100 nM - 100 µM; we chose the greatest concentration in which the drug itself did not have an effect on cell viability or caspase activation (data not shown).
Determination of Cell Viability
The percentage of live cells was determined by means of the Trypan blue exclusion technique. In brief, once treatments were completed cells were rinsed in phosphate buffered saline (PBS) and resuspended with trypsin/EDTA; after trypsinisation was stopped, cells were immediately stained with 0.5% Trypan blue; the number of live and dead cells were determined in a Neubauer chamber. The results are expressed as the percentage of live cells compared to the control. We performed a minimum of 5 independent experiments for each of the analyses.
Preparation of Cell Extracts and Analysis by Western Blot
After treatments were completed, cells were washed, recovered and suspended in Tris-HCl buffer 10 mM pH 7.4, 1% Triton X-100 and 0.1 mM PMSF. The suspension was centrifuged for 20 min at 10 000 g and the protein content of the supernatant was determined by the bicinchoninic acid method. To determine the phosphorylation of SAPK/JNK, ERK1/2, and p-38 MAPK, cardiocytes were lysed in 50 µl of Tris-HCl buffer 10 mM pH 7.4, 1 mM PMSF, 1% Igepal CA-630 and 1% phosphate inhibitor cocktail.
The activation of caspases 3, 8, and 9 and the phosphorylation of SAPK/JNK, ERK1/2, and p-38 MAPK were determined by Western blot. We separated 20 µg of each protein extract by SDS-PAGE and transferred it to PVDF membranes (Millipore, Bedford, MA, USA). These were blocked with 5% non-fat milk in PBS and incubated overnight at 4ºC in binding buffer (PBS, Tween 20 0.1%) with the primary antibody for each protein (1:1000). The next day the membranes were washed and incubated for 60 min at room temperature with the secondary antibody marked with peroxidase (1:5000). After washing again, the binding of the antibody was detected by chemiluminescence with ECL (GE Healthcare Biosciences, Netherlands). We performed a minimum of 4 experiments for each of the proteins analyzed. The quantitative analysis was performed with the Gel Pro Analyzer 3.1 program (Sigma).
Determination of DNA Fragmentation
For DNA extraction, cells were lysed in Tris-HCl buffer 50 mM pH 7.4, EDTA 20 mM and Igepal CA-630 1% and incubated, first in SDS 2% and RNasa 5 µg/µL for 2 h at 56ºC, then in proteinase K 2.5 µg/µL for 2h at 37ºC. Subsequently, the DNA was precipitated in ethanol and finally resuspended in distilled water. The fragmentation of the DNA was analyzed by electrophoretic separation in 2% agarose gels, which were then stained with ethidium bromide 0.2 µg/µL.
Statistical Analysis
The results are shown as mean (standard deviation) for a number (n) of independent experiments. The differences between groups were analyzed with the non-parametric Mann-Whitney U test. We considered values of P<.05 as significant.
RESULTS
Apoptosis and Testosterone
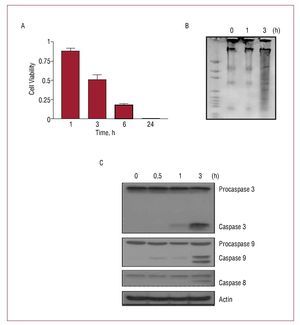
The induction of hyperosmotic stress in H9c2 cells by treatment with the non-permeable solute sorbitol (0.6 M) produced a rapid and time-dependent decrease in cell viability, an increment in the fragmentation of DNA and an activation of the proteolytic caspases 3, 8, and 9 (Figure 1). The length of sorbitol treatment selected to induce apoptosis in the following tests was 3 hours, the time at which of the above-named effects were significant.

Figure 1. Hyperosmotic stress induced a rapid and time-dependent apoptosis in H9c2 cells. Cells were incubated for different lengths of time in sorbitol 0.6 M in a culture medium free of plasma and antibiotics. (A) Cell viability. Values are mean (standard deviation) of 5 independent experiments. (B) Activation of the fragmentation of DNA and (C) activation of caspases 3, 8 and 9, analyzed by Western blot as a response to the presence of sorbitol.
The effects of co-treatment of sorbitol with testosterone and/or flutamide on cell viability and activation of the caspases is shown in Figure 2. Note that the administration of testosterone (T) or flutamide (F) in the absence of sorbitol did not produce any effect. Compared to the treatment with sorbitol alone (S), the co-treatment with testosterone was associated with an increment in cell viability (S+T vs S, P<.001). The further addition of flutamide, an antagonist of the androgen receptor, did not alter this protector effect of testosterone (S+T+F vs S+T, P=NS). Similarly, the co-treatment with testosterone reduced the activation induced by sorbitol in all the analyzed caspases (S+T vs. S, P<.01), and the addition of flutamide increased this inhibition (S+T+F vs S+T, P<.05).

Figure 2. Effects of testosterone on the apoptosis induced by sorbitol. (A) Cell viability. (B) Western blot of the activation of the caspases and (C-E) densitometry analysis calculated as active caspase/ total procaspase for caspases 3 and 9, and as active caspase/actin for caspase 8. Results are shown as mean ± standard deviation of 6 independent experiments. Control (C); flutamide (F), sorbitol (S), testosterone (T). aP<.001 with respect to sorbitol. bP<.001 with respect to sorbitol + testosterone. cP<.01 with respect to sorbitol. dP<.05 with respect to sorbitol + testosterone
Figure 3 shows the results of the analysis of the protein kinases activated by mitogens (MAPK) involved in apoptosis induced by sorbitol. The treatment with sorbitol alone (1 h) induced the phosphorylation of SAPK/JNK and decreased the phosphorylation of ERK1/2, but had no effect on p38 MAPK (data not shown). The co-administration of testosterone significantly reverted the effects of sorbitol, decreasing the activation of SAPK/JNK and increasing the activation of ERK1/2. Again, these effects of testosterone were not altered by the presence of flutamide.

Fig. 3. Effects of testosterone on the signal cascades of the MAPK. Western blot and densitometry analysis of the activation of (A) JNK and (B) ERK 1/2. After 1 h of treatment, the activation of the kinases was calculated as phosphorylated protein/ total protein. Results are shown as mean (standard deviation) (n=4 independent experiments). Control (C); flutamide (F), sorbitol (S), testosterone (T). aP<.01 bP<.001 with respect to sorbitol (S).
Testosterone, Spironolactone, and Eplerenone
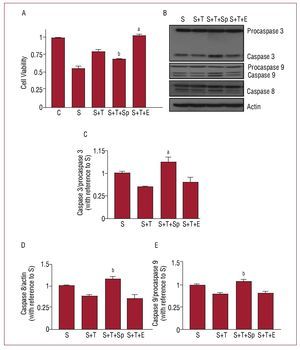
Figure 4 shows the effects of spironolactone and eplerenone on the decrease of apoptosis mediated by testosterone. The administration of spironolactone or eplerenone had no effect on cell viability or activation of the caspases in H9c2 cells, nor did it produce any change in the apoptosis induced by sorbitol (data not shown). However, spironolactone and eplerenone had opposite effects on the protective role of testosterone. The pre-treatment with spironolactone blocked the effects of testosterone, reducing cell viability (S+T+Sp vs S+T, P<.01) and increasing the activation of caspases 3, 8, and 9 (S+T+Sp vs S+T, P<.01). By contrast, pre-treatment with eplerenone increased cell viability (S+T+Ep vs S+T, P<.001) but did not affect the decrease of caspase activation induced by testosterone (S+T+Ep vs S+T, P=NS).

Figure 4. Effects of spironolactone and eplerenone on cell viability and caspase activation. (A) Cell viability. (B) Western blot of the activity of the caspases and (C-E) densitometry analysis for each caspase. Results are shown as mean ± standard deviation of 5 independent experiments. Control (C); eplerenone (E); flutamide (F), sorbitol (S), spironolactone (Sp); testosterone (T). aP<.001 with respect to sorbitol + testosterone. bP<.01 with respect to sorbitol + testosterone.
DISCUSSION
The main findings of this study are the following: a) testosterone reduces apoptosis induced by hyperosmotic stress in cardiomyocyte strain H9c2; b) this action is not mediated by the androgen receptor, and involves the SAPK/JNK and ERK 1/2 signalling cascades; and c) this protective effect of testosterone is antagonized by spironolactone but not by eplerenone, suggesting differential effects of these drugs.
In this study we used hyperosmotic stress as an inductor of apoptosis because it is one of the principal mechanisms of tissue damage in pathological states such as ischaemia, septic shock and acidosis.14 In addition, it is a model of apoptosis induction which is more rapid and potent than other classical models such as hypoxia, doxorubicin or angiotensin II,15 which allowed us to investigate the possible non-genomic effects of testosterone on induced apoptosis. This is the first study to demonstrate that testosterone, by means of a rapid, non-genomic route, protects H9c2 cardioblasts from the apoptosis induced by hyperosmotic stress. This hypothesis was confirmed by the fact that flutamide, a blocker of androgen receptors, did not block the protective effect of testosterone. However, the mechanisms of this beneficial effect of testosterone still need to be determined.
It is well established that the classical pathway of action of testosterone implies its binding to the androgen receptor and the translocation of the testosterone-receptor complex to the nucleus, which finally activates protein synthesis.16 Recently a rapid and non-genomic action of testosterone has been demonstrated in cardiocytes17,18 and other cell systems,6 independent of the androgen receptor. Thus, by means of its binding to a membrane g protein-coupled receptor (GPCR), testosterone is capable of activating the cascade PLC/I3P and inducing a rapid intracellular increase in calcium in rat cardiocyte primary culture.18 In skeletal muscle cells, by means of its binding to a GPCR and by increment of intracellular calcium, testosterone is capable of activating the signalization route MEK/ ERK and mediating an anti-apoptotic effect, similar to that obtained in our results. On the other hand, it has been demonstrated that testosterone is a strong blocker of the type L calcium channels,19 which participate in the activation of apoptosis in a number of cell systems including cardiocytes.20 Our results demonstrate that the ERK1/2 and SAPK/JNK cascades are involved in the inhibition of apoptosis mediated by testosterone. However, further investigation is necessary to define if this protective effect is due to the blockage of the type L calcium channels or to the increment of intracellular calcium by means of its binding to a GPCR. It is worth mentioning that other groups have previously demonstrated a pro-apoptotic effect of testosterone in cardiac cells,21 however, in these studies the adverse effect of testosterone was mediated by long expositions (≥20 h) to the hormone, suggesting a genomic route in accordance with the classical route of testosterone by means of its binding to the androgen receptor, different from the rapid effect of testosterone described here. Similarly, it is known that androgens mediate protective effects due to the catabolism induced by glucocorticoids.22 The glucocorticoid receptors, like the androgen receptor, are cytoplasmic receptors which, when they bind with their ligand, are translocated to the nucleus where they finally activate protein synthesis. This is a slow mechanism from the binding with the ligand to the expression of proteins. If the anti-apoptotic effect of testosterone were due to its capacity to block an apoptotic effect mediated by the glucocorticoids, this protector effect would not be seen until a much later time (24 hours) than that described in our results. This fact allows us to discount the glucocorticoid receptor as a possible target to explain the rapid non-genomic effects described in this study.
Spironolactone and eplerenone are 2 drugs habitually used in the treatment of heart failure,23 which have significant structural differences. Spironolactone is an antimineralocorticoid associated with progestogenic and anti-androgenic effects,11 while eplerenone is a derivative of spironolactone designed to increase its selectivity to the mineralocorticoid receptor while minimizing its progestogenic and anti-androgenic effects.11,12 In our study spironolactone reduced the protective effects of testosterone, while eplerenone had no blocking effect. This differential action of the 2 drugs cannot be attributed to their different capacities of binding to the androgen receptor, since the protective effect of testosterone was independent of this receptor. The differences may be due to the progestogenic effects present in spironolactone and absent in eplerenone. It has been shown that progesterone mediates non-genomic effects in different types of tissues,24 including the modification of calcium flow,25 levels of AMPc26 and MAPK signalling.27 As a result, by having structural elements of the progesterone molecule and progestogenic effects,11 spironolactone appears to antagonize the anti-apoptotic action of testosterone. Eplerenone, whose progestogenic effects have been eliminated, does not appear to antagonize the protector effect of testosterone. There are currently no studies which demonstrate this hypothesis, only a few in which spironolactone induced rapid increments in the intracellular calcium of the myocardium,28 which did not investigate the mechanisms implicated. Our results thus suggest the need for further studies which investigate the mechanisms induced by spironolactone, not present in eplerenone, which are implicated in the blockage of the beneficial effects of testosterone.
Clinical Implications
The findings of this study may have important clinical implications in terms of apoptosis being a mechanism of cell death in HF, in which it has also been demonstrated that a deficit of testosterone is associated with a poorer prognosis.2-4 Our results suggest that the anabolic deficit present in advanced stages of HF may be a determining factor in the greater degree of apoptosis described in this illness, and thus in a poorer clinical evolution. At present, the treatment of HF with testosterone is an attractive therapeutic option being evaluated, which has been shown to improve the symptoms, the capacity for exercise and the quality of life in a few tests with a small number of patients.2,29 However, its effect on the evaluation of the illness has not yet been evaluated.29
On the other hand, it is known that spironolactone and eplerenone are two drugs of common use in advanced HF due to their beneficial effects as blockers of the aldosterone receptor. The differences between these drugs in terms of a lower rate of secondary effects produced by eplerenone for its low anti-androgenic action are well known. However, up to now no differences in their action at the cardiac level have been described, nor are there direct comparative studies of these two drugs. Our study is the first to show the differences in selectivity of spironolactone and eplerenone on the action of testosterone at the level of the cardiocyte. Our findings suggest that the benefits of spironolactone on the cardiomyocyte may be diminished by blocking the protective effect of testosterone. By contrast, eplerenone may have an advantage with respect to spironolactone, since it does not diminish the anti-apoptotic effect of testosterone. However, further basic and clinical studies are needed to evaluate this possibility in in vivo models.
Limitations
The cardiocytes used in this study were from the H9c2 embryonic cell line of cardiac myoblasts, which may have phenotypic differences with adult cardiocytes. Spironolactone and eplerenone were used at concentrations that, although similar to those of other in vitro studies, are not comparable to those habitually used in clinical practice, and we did not perform a dose-response study. Therefore, the results presented in this study should be confirmed in future studies using in vivo models, using concentrations comparable to those of clinical practice. These HF models should preferably be chronic and take into account the possible interactions with other types of cells such as fibroblasts, as well as the phenomena of fibrosis and remodelling, not evaluated in this in vitro study.
CONCLUSIONS
This study showed that treatment with testosterone protects the H9c2 embryonic cardiac cell line from the apoptosis induced by hyperosmotic stress, by non-genomic means independent of the androgen receptor in which at least SAPK/JNK and ERK1/2 are involved. Furthermore, this beneficial effect of testosterone is blocked by spironolactone but not by eplerenone, which suggests a differential action of the two drugs and a possible additional benefit of eplerenone compared with spironolactone.
ABBREVIATIONS
GPCR: G protein-coupled receptor
HF: heart failure
MAPK: mitogen-activated protein kinase
SEE EDITORIAL ON PAGES 760-2
This study was partially financed by a fellowship of the Seneca Foundation (Science and Technology Agency of the Region of Murcia) [05822/PPC/07], by the Spanish national research network for heart failure "REDINSCOR" (Spanish Ministry of Health and Consumer Affairs) [RD06/0003/0013] and by a grant from Pfizer Inc., NY, USA. We thank Prof F. Fernández-Belda (Faculty of Veterinary, Murcia, Spain) for generously supplying the cell line H9c2.
Correspondence: Dr. D.A. Pascual Figal.
Unidad de Insuficiencia Cardiaca. Servicio de Cardiología. Hospital Universitario Virgen de la Arrixaca.
Ctra. Madrid-Cartagena, s/n. 30120 El Palmar. Murcia. España.
E-mail: dapascual@servicam.com
Received November 21, 2009.
Accepted for publication January 25, 2010.