
Existen diferentes modelos de aterosclerosis en animales tanto pequeños (roedores) como grandes. La gran ventaja de los animales pequeños es que son de bajo coste, asequibles y de fácil manejo y que hay disponibles modificaciones genéticas relevantes. Los modelos de aterosclerosis en animal grande tienen las desventajas de sus peores asequibilidad y manejo y su coste. Sin embargo, tienen anatomía y fisiología mucho más parecidas a las humanas y los resultados obtenidos son mucho más extrapolables a la patología humana. La elección de uno u otro modelo depende de la naturaleza que se quiera dar a la investigación. En estudios mecanísticos, se puede utilizar con mayor precisión los modelos de roedor, pero para un estudio más traslacional, los modelos de animal grande dan respuestas más cercanas a las de los pacientes. Las técnicas de imagen no invasiva han hecho avanzar mucho el campo del conocimiento de la aterosclerosis a través del estudio seriado de modelos animales ateroscleróticos.
Palabras clave
Ignatowski1 aportó la primera evidencia de aterosclerosis experimental cuando, en 1908, describió que, al alimentar a conejos con dieta rica en proteínas animales (carne, leche, huevos, etc.), se inducía la formación de células (macrófagos) grandes y claras. Sin embargo, fue Anitschkow2 el primero en demostrar que la inducción de la aterosclerosis se debía a los lípidos, no a las proteínas; alimentó conejos con yema de huevo y colesterol disuelto en aceite vegetal y comprobó que se producían lesiones ateroscleróticas similares a las presentes en humanos. Desde estos estudios pioneros hasta la actualidad, se ha recorrido un largo camino respecto a los modelos animales de aterosclerosis. En el presente artículo se resumen sucintamente las consideraciones más interesantes acerca de los modelos animales.
Proceso de AterogénesisLa acumulación de colesterol tiene un papel central en el proceso de aterogénesis. La primera etapa consiste en que, a causa de los factores de riesgo, el endotelio aumenta su permeabilidad (disfunción endotelial, el fenómeno más precoz de la aterogénesis). El colesterol unido a lipoproteínas de baja densidad (cLDL) penetra en la pared vascular merced al proceso de disfunción endotelial, se une a los proteoglucanos del espacio subendotelial y sufre oxidación. El colesterol oxidado es altamente tóxico, de modo que es fagocitado por los macrófagos de la pared arterial; la presencia de colesterol oxidado desencadena una serie de reacciones proinflamatorias a través de diversos mediadores que perpetúan la inflamación y reclutan más monocitos/macrófagos y células inflamatorias. Al fagocitar los lípidos, los macrófagos se transforman en células espumosas. Se producen cambios secundarios en la media y en la adventicia, con proliferación de células de músculo liso vascular (CML) y aparición de una cubierta fibrótica que protege el núcleo lipídico de la sangre. Cuando los macrófagos no pueden acumular más colesterol, inician el proceso de apoptosis, y liberan no solo el colesterol nuevamente al espacio subendotelial, sino además sustancias proinflamatorias como factor tisular, citocinas y metaloproteinasas de matriz (las cuales digieren la cubierta fibrosa), lo cual hace a las lesiones más susceptibles a la erosión y la rotura. Cuando la placa se rompe, la sangre entra en contacto con el núcleo lipídico necrótico (altamente trombogénico), lo cual induce la formación de un trombo sobre esa placa, trombo que ocluye la circulación obstruyendo por completo la luz del vaso, lo que desencadena un evento cardiovascular (síndrome coronario agudo o ictus).
Modelos de Aterosclerosis en RatonesClásicamente los estudios con modelos animales se habían llevado a cabo con conejos, cerdos y primates. No obstante, las dificultades de manejo de animales grandes, los gastos que conllevan dichos animales y las consideraciones de carácter ético al usar primates condicionaron el desarrollo de modelos de aterosclerosis en animales más pequeños. Además, el desarrollo tecnológico, con la aparición de ratones modificados genéticamente, hizo que este modelo se extendiera rápidamente.
Ratones SilvestresLos ratones no modificados genéticamente se denominan ratones silvestres (del inglés wild type mouse). En los ratones silvestres no se desarrolla espontáneamente la aterosclerosis cuando se los alimenta con la dieta habitual del ratón. Por ello, para iniciar el proceso aterogénico, se necesita elevar la concentración de LDL (cociente HDL/LDL bajo). Esto se consigue con dietas ricas en colesterol.
Los estudios iniciales en ratones se hicieron con dietas hipercolesterolémicas en diversas razas silvestres. El pionero fue Robert Wissler, que usó ácidos biliares para inducir la formación de placas3. Una de las más populares es la dieta Paigen (desarrollada en 1985 por Beverly Paigen4,5), que es una dieta rica en grasa (el 15%, principalmente grasas saturadas derivadas de mantequilla de coco, se suele añadir un 1% de aceite de maíz para suplir la deficiencia de ácidos grasos poliinsaturados) y en colesterol (1,25%), que además contiene ácido cólico (0,5%). El desarrollo de estría grasa es proporcional a la composición en grasas saturadas. El ácido cólico actúa aumentando la absorción de colesterol (como sal biliar que es) e inhibiendo la excreción de ácidos biliares (inhibe la enzima 7α-hidroxilasa); no obstante, modernos estudios recomiendan no emplear ácido cólico, pues activa genes implicados en fibrosis hepática6.
Paigen estudió la susceptibilidad a la aterosclerosis de diez razas diferentes de ratón tratadas con la dieta Paigen; de menor a mayor susceptibilidad, encontramos: BALB/cJ = C3H/J = A/J = SWR/J = NZB/J (apenas desarrollan) < 129J = = AKR/J = DBA/2J < C57L/J < C57BL/6J. La dieta indujo hipercolesterolemia en las diez razas, pero solo aterosclerosis en las cinco últimas; no hubo relación estadística entre hipercolesterolemia y desarrollo de aterosclerosis. Además, incluso en la raza C57BL/6J (la más susceptible) las lesiones se restringían al arco aórtico, eran pequeñas y contenían pocas células.
Por este motivo, se desarrollaron modelos de ratón modificados genéticamente; los primeros en desarrollarse fueron los de ratón knock out (KO) para apoE y ratón KO para LDL-R. Se denomina KO a la ausencia del gen en cuestión. Debemos destacar que ningún modelo murino disponible desarrolla todo el espectro de lesiones presentes en humanos. La mayor parte de los modelos de ratón desarrollan lesiones tipo I y II de la clasificación de la American Heart Association (AHA), pero solo un número muy escaso desarrolla lesiones tipo IV. Ningún modelo de ratón desarrolla la rotura o la erosión de placa ni la trombosis que causan los eventos cardiovasculares en humanos. Por ellos, se debe usar los modelos murinos para estudiar depósito lipídico y adhesión de monocitos.
Ratones KO para apoEEn 1992, los laboratorios de Breslow7 y Maeda8, trabajando independientemente, comunicaron a la comunidad científica que habían desarrollado el modelo de ratón KO para apoE, en el que las lesiones ateroscleróticas eran muy frecuentes (incluso con dietas normales). A diferencia del ratón silvestre (que solo sufre lesiones —estría lipídica— en la raíz aórtica y con dieta rica en colesterol), el ratón KO para apoE adquiere lesiones en todo el árbol arterial9, incluso con dieta normal, y dichas lesiones progresan más allá de la estría grasa hasta la placa fibrosa.
En el ratón KO para apoE, una dieta tan agresiva como la Paigen no es necesaria (de hecho, este modelo adquiere espontáneamente lesiones incluso con dieta normal). No obstante, una dieta rica en colesterol acelera el desarrollo de la placa y la vuelve aún más rica en lípidos. La dieta más usada en todos los experimentos con ratones es la occidental (Western diet, introducida por Plump en el laboratorio de Breslow en 1992)7, que contiene un 21% de grasa y el 0,15% de colesterol; de hecho, se ha demostrado que la dieta occidental en ratones KO para apoE acelera el desarrollo de la placa sin cambiar las características morfológicas de la lesión9.
Los ratones heterocigotos para apoE (ApoE–/+) no desarrollan aterosclerosis con dieta normal, pero sí con dieta rica en colesterol; sin embargo, el ratón homocigoto ApoE–/– sí es significativamente hiperlipémico (2.000mg/dl) y sufre lesiones 50 veces más extensas que el heterocigoto ApoE–/+.
Ratones KO para el receptor de LDL (LDL-R)El otro modelo murino más usado es el de ratón KO para LDL-R, creado en el laboratorio de Herz en 199310. Comparado con los ratones silvestres, los KO para LDL-R presentan altas concentraciones de cLDL (fenotipo muy similar al de los humanos con hipercolesterolemia familiar), pero con dieta normal las lesiones ateroscleróticas se desarrollan muy lentamente. No obstante, al alimentarlos con dieta rica en colesterol incrementan los valores de LDL y lipoproteínas de muy baja densidad (VLDL) (se alcanza el nuevo equilibrio lipídico en 2 semanas) y las lesiones se desarrollan en 2–3 meses. La dieta más usada en este modelo es la dieta occidental (el 21% de grasa, el 0,15% colesterol, idealmente suplementada con un 1% de aceite de maíz para obtener ácidos grasos esenciales); de hecho, se ha demostrado que la dieta occidental en ratones KO para apoE acelera el desarrollo de la placa sin cambiar las características morfológicas de la lesión9.
Tanto el ratón KO para apoE como el KO para LDL-R son modelos hiperlipémicos, pero los primeros se caracterizan por sufrir lesiones más graves o más complejas que los segundos y porque las lesiones aparecen incluso con dieta normal11,12.
Algunos investigadores prefieren el modelo de ratones KO para LDL-R a pesar de sus lesiones más pequeñas y que necesitan dietas especiales, dado que la distribución del colesterol en estos ratones se parece más a la de los humanos, en que la mayor parte del colesterol se moviliza en cLDL. El colesterol en los ratones KO para apoE se moviliza principalmente en forma de VLDL.
Modelo de regresión de lesiones en el ratónEs importante distinguir entre retraso en la progresión de las lesiones y regresión auténtica. Existen dos modelos murinos de regresión de lesiones ya establecidas:
- •
Trasplante de aorta: consiste en trasplantar un segmento de aorta de un ratón KO para apoE a un ratón normal (100mg/dl). El tamaño de las lesiones trasplantadas se reduce un 50% en unos 3 meses.
- •
Ratón apoE/Mx1-Cre: estos ratones tienen colesterol normal con dieta normal; cuando se les da un dieta con el 16% grasa, el 1,25% de colesterol y el 0,5% de ácido cólico, el colesterol aumenta 1.000mg/dl.
Consiste en trasplantar parte de la aorta de ratones KO para apoE –/– (colesterol, unos 600mg/dl) a ratones C57BL/6 con valores lipídicos normales (< 100mg/dl) (tablas 1 y 2). Ventajas de usar un modelo de ratón: facilidad en el manejo por pequeño tamaño; son baratos (tanto la compra inicial como el mantenimiento); la rapidez en el desarrollo de lesiones. Inconvenientes de usar un modelo de ratón: existen marcadas diferencias entre la placa humana y la del ratón (en este no se dan rotura de placa ni trombosis); el perfil lipídico del ratón (especialmente el KO para apoE, que transporta la mayor parte del colesterol en VLDL) es marcadamente diferente del humano (que transporta la mayor parte del colesterol en LDL); las arterias del ratón son de muy pequeño tamaño, con las consiguientes diferencias en morfología de la pared arterial respecto a los humanos, como una túnica media de espesor mucho menor y falta de vasa vasorum; ciertos procedimientos, como la denudación endotelial mediante balón, no han sido exitosos en los vasos murinos.
Modelos de ratón genéticamente modificados empleados para el desarrollo de aterosclerosis.
| Ratón | Características de la lesión | Localización de la lesión | Requiere dieta especial | Disponibilidad comercial |
| Deleciones | ||||
| ApoE–/–7,8 | Progresa desde células espumosas hasta núcleos necróticos con cápsula fibrosa | Raíz aórtica, aorta, carótida | No | Jackson |
| ApoE–/+62 | Células espumosas sobrecargadas de lípidos | Raíz aórtica | Sí | Taconic |
| LDL-R–/–10 | Células espumosas sobrecargadas de lípidos | Raíz aórtica, aorta | Sí | Jackson |
| Transgénicos | ||||
| ApoB–63 | Células espumosas sobrecargadas de lípidos | Raíz aórtica | Sí | Taconic |
| Apo(a)64 | Células espumosas sobrecargadas de lípidos | Raíz aórtica | Sí | |
| CETP65 | Lesiones ricas en lípidos | Raíz aórtica | Sí | Jackson |
| ApoE(arg112, Cys142)66 | Células espumosas sobrecargadas de lípidos | Raíz aórtica | Sí | |
| ApoE3Leiden67 | Células espumosas sobrecargadas de lípidos | Raíz aórtica | Sí | |
| ApoC-III-68 | No definidas | Raíz aórtica | Sí | |
| Compuestos | Raíz aórtica | Sí | ||
| Apobec–/– x LDL-R -/-69 | Varía de lesiones con células espumosas a fibrosis e hiperplasia de CML | Raíz aórtica | ||
Apo: apolioproteína; CETP: proteína de transferencia de ésteres de colesterol; CML: células de músculo liso vascular; LDL-R: receptor de lipoproteínas de baja densidad.
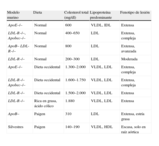
Fenotipo lipídico de los modelos murinos de aterosclerosis.
| Modelo murino | Dieta | Colesterol total (mg/dl) | Lipoproteína predominante | Fenotipo de lesión |
| ApoE–/– | Normal | 600 | VLDL, IDL | Extensa |
| LDL-R–/–, Apobec–/– | Normal | 400–650 | LDL | Extensa, compleja |
| ApoB– LDL-R–/– | Normal | 800 | LDL | Extensa, avanzada |
| LDL-R–/– | Normal | 200–300 | LDL | Moderada |
| ApoE–/– | Dieta occidental | 1.300–2.000 | VLDL, LDL | Extensa, compleja |
| LDL-R–/– Apobec–/– | Dieta occidental | 1.600–1.750 | VLDL, LDL | Extensa, compleja |
| LDL-R–/– | Dieta occidental | 1.500–2.000 | VLDL, LDL | Extensa |
| LDL-R–/– | Rica en grasa, ácido cólico | 1.880 | VLDL, LDL | Extensa |
| ApoB– | Paigen | 310 | LDL | Extensa, estría grasa |
| Silvestres | Paigen | 140–190 | VLDL, HDL | Escasa, solo en raíz aórtica |
Apo: apolioproteína; LDL-R: receptor de lipoproteínas de baja densidad.
La rata es una especie bastante resistente a la aterosclerosis. Igual que el ratón y a diferencia de los humanos, no expresa proteína de transferencia de ésteres de colesterol (CETP) y el colesterol se transporta en su mayor parte en forma de cHDL. Las ratas generalmente responden poco al colesterol de la dieta, por ello la aterogénesis solo puede inducirse (y en escasa cuantía) con dietas muy ricas en colesterol y grasas que contengan además ácido cólico y tiouracilo. El ácido cólico actúa incrementando la absorción de colesterol y suprimiendo la actividad de 7α-hidroxilasa (la enzima limitante en la síntesis de ácidos biliares, es decir, en la excreción del colesterol); el tiouracilo actúa induciendo hipotiroidismo clínico, que reduce el número y la actividad de LDL-R hepáticos (lo que conduce a un incremento en la concentración de LDL).
Modelos de Aterosclerosis en HámsterEn el hámster se desarrollan hipercolesterolemia y aterosclerosis precoz con dietas aterogénicas. Por ejemplo, una dieta de 2 meses con el 0,2% de colesterol y el 10% de aceite de coco produjo en el hámster sirio dorado macho13 incremento del colesterol total en un 400%, las VLDL/lipoproteínas de densidad intermedia (IDL) del 200% y las LDL el 20%, y descenso del 45% en las HDL, así como formación de estría grasa selectivamente en la aorta ascendente. Pese a ello, actualmente no son modelos de uso muy extendido.
Modelos de Aterosclerosis en ConejosLos conejos son un modelo de aterosclerosis usado muy frecuentemente. El conejo más ampliamente usado es el New Zealand White (NZW). En el conejo se desarrolla aterosclerosis avanzada, pero se necesita hipercolesterolemia marcada; dado que el conejo absorbe colesterol de manera muy eficiente, se puede inducir estas altas concentraciones de colesterol mediante dieta rica en colesterol.
Ventajas: modelo barato, fácilmente disponible, de fácil manejo, no tiene requerimientos especiales y el perfil lipoproteico es relativamente similar al humano (excepto por deficiencia en lipasa hepática). Inconvenientes: se requiere una hiperlipemia extrema para producir las lesiones, esto produce acumulación de colesterol en corazón, riñones, hígado y pulmones (órganos no afectados por aterosclerosis en humanos); incluso el hígado graso así inducido puede llegar a causar fallo hepático con ictericia, prurito, etc., e incluso muerte durante la fase de inducción.
Modos de inducción de aterosclerosis en conejoDietaLas dietas usadas contienen habitualmente entre el 0,5 y el 2% de colesterol y se administran durante un tiempo variable de 4–16 semanas, dependiendo de la gravedad de la lesión que se quiera alcanzar. A corto plazo, la dieta es relativamente bien tolerada por los conejos, aunque produce gran variedad de tamaños de las lesiones. Las lesiones se producen principalmente en el arco aórtico y la aorta ascendente, y habitualmente no en la aorta abdominal.
El conejo es el único animal que tiene tendencia a sufrir hipercolesterolemia a los pocos días de administración de dieta rica en colesterol (en los primeros 20 días, la concentración se incrementa 2–8 veces)14. En condiciones de hipercolesterolemia, se forman rápidamente las primeras lesiones (células espumosas, similares a las humanas). Sin embargo, este modelo no se puede mantener a largo plazo, pues la hepatotoxicidad de la dieta acarrea una muy alta mortalidad; además, se trata de una situación no fisiológica, pues la inflamación masiva a medio plazo que se produce en los conejos con dieta muy rica en colesterol a largo plazo no se asemeja a las condiciones en que se desarrolla la aterosclerosis humana (inflamación de bajo grado pero persistente). No obstante, algunos investigadores utilizan este modelo para el estudio de los efectos de los fármacos en la estría grasa; de hecho, nuestro grupo utilizó este modelo para demostrar por primera vez que es posible conseguir la regresión de estría grasa mediante la administración de cHDL purificado de conejo15,16.
Dieta y denudación aórticaEs el método que ofrece unas lesiones más parecidas a las humanas, motivo por el cual está ampliamente establecido en todo el mundo y es el empleado en nuestro laboratorio17–29. La lesión (única o doble) con el balón de angioplastia produce denudación de la aorta, lo cual acelera el proceso de formación de lesiones ateroscléroticas y genera unas placas con un núcleo lipídico rodeado por una cubierta fibrosa con gran proliferación de CML. De hecho, estas lesiones son más similares a las humanas que las producidas en conejos solo con dieta rica en colesterol30,31.
Brevemente, el método consiste en inducir lesiones ateroscleróticas aórticas mediante la combinación de 9 meses de dieta rica en colesterol (0,2%) y doble denudación aórtica. La denudación aórtica se lleva a cabo a los 30 días y nuevamente a los 90 días de empezar la dieta rica en colesterol; se realiza una disección anatómica de la región inguinal para exponer la arteria femoral, se canula la arteria femoral, con guía fluoroscópica se sube un balón Fogarty de 4 Fr hasta el arco aórtico, se infla el balón y se retira suavemente, con lo que se produce la denudación del endotelio de la aorta desde el arco aórtico hasta la iliaca. Las lesiones así generadas son muy similares a las humanas, pues no solamente presentan células espumosas, sino además importante componente de fibrosis y proliferación de CML.
Variantes genéticasEl modelo principal es el conejo Watanabe con hiperlipemia heredable (WHHL). En 1980, Watanabe reportó una cepa creada a partir de un único conejo hipercolesterolémico32. Actualmente se sabe que los conejos Watanabe tienen un defecto en LDL-R; es decir, un modelo que remeda la hipercolesterolemia familiar (hiperlipoproteinemia hereditaria tipo IIa de Frederickson). Desde entonces, es uno de los modelos más estudiados, tanto para el metabolismo de lipoproteínas como para el desarrollo de aterosclerosis o el efecto de diversos fármacos (principalmente estatinas). Aparecen lesiones en todos los estadios de progresión, desde estría lipídica a placas avanzadas. Las lesiones se concentran en las coronarias y la aorta, y los lípidos se acumulan tanto en las células espumosas como en CML.
En los conejos Watanabe se desarrolla una doble alteración: a) deficiencia de LDL-R, que produce hipercolesterolemia, y b) altas concentraciones de VLDL y bajas de HDL, junto con acumulación de grasa visceral e hiperinsulinemia, que remedan el síndrome metabólico humano. Esto causa que se desarrolle tanto aterosclerosis como infarto agudo de miocardio (IAM), alteraciones valvulares, xantomas33, etc. Las principales diferencias en el metabolismo lipídico entre los conejos Watanabe, los humanos con hipercolesterolemia familiar y los ratones KO para apoE y LDL-R se resumen en la tabla 3.
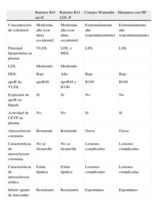
Diferencias en el metabolismo lipídico y sus complicaciones entre humanos con hipercolesterolemia familiar (HF), conejo Watanabe y ratones knock out para apoE y LDL-R.
| Ratones KO apoE | Ratones KO LDL-R | Conejos Watanabe | Humanos con HF | |
| Concentración de colesterol | Moderada-alta (con dieta occidental) | Moderada-alta (con dieta occidental) | Extremadamente alta (espontáneamente) | Extremadamente alta (espontáneamente) |
| Principal lipoproteína en plasma | VLDL | LDL y HDL | LDL | LDL |
| LDL | Moderado | Moderado | ||
| HDL | Bajo | Alto | Bajo | Bajo |
| apoB de VLDL | apoB48 | apoB48 y B100 | B100 | B100 |
| Expresión de apoB en hígado | Sí | Sí | No | No |
| Actividad de CETP en plasma | No | No | Sí | Sí |
| Aterosclerosis coronaria | Resistente | Resistente | Grave | Grave |
| Características de aterosclerosis coronaria | No se desarrolla | No se desarrolla | Lesiones complicadas | Lesiones complicadas |
| Características de aterosclerosis aórtica | Estría lipídica | Estría lipídica | Lesiones complicadas | Lesiones complicadas |
| Infarto agudo de miocardio | Resistentes | Resistentes | Espontáneo | Espontáneo |
CETP: proteína de transferencia de ésteres de colesterol; HDL: lipoproteínas de alta densidad; HF: hipercolesterolemia familiar; KO: knock out; LDL: lipoproteínas de baja densidad; LDL-R: receptor de lipoproteínas de baja densidad; VLDL: lipoproteínas de muy baja densidad.
No obstante, en los conejos descritos inicialmente por Watanabe se desarrollaba aterosclerosis aórtica, pero no coronaria. Con cría selectiva se desarrollaron conejos que sufren aterosclerosis coronaria34. Otra modificación posterior es el desarrollo de conejos WHHL propensos al IAM (WHHLMI)35, en los que se observa aterosclerosis coronaria ya a los 2 meses; a los 20 meses, estenosis del 70%; a los 20 meses, estenosis del 90%, y a los 30 meses, IAM en el 97% de los casos, con lesiones muy similares a las humanas y calcificación y hemorragia en la placa.
La raza del Hospital St. Thomas (STH) tiene un perfil lipídico similar al de los pacientes que sufren hipertrigliceridemia e hiperlipemia combinada, y asimismo contrae aterosclerosis36,37.
La morfología de la placa se ve influida por la composición y la duración de la dieta38. Dietas de corta duración y más del 2% de colesterol en la dieta causan lesiones ricas en células espumosas. Dietas con bajo porcentaje de colesterol (0,1–0,2%) pero mantenidas durante largo tiempo originan lesionas más similares las humanas, con mayor porcentaje de tejido fibroadiposo14. Con un aporte superior al 0,15%, ya se detectan ésteres de colesterol en la placa.
La localización de las lesiones en el modelo de lesión + dieta es selectivo en el punto de la aorta donde se produce la denudación endotelial (casi siempre en la aorta descendente). En el modelo de dieta, las lesiones se localizan en arco aórtico, la aorta torácica en la salida de las arterias intercostales y, en menor cuantía, en la aorta abdominal.
Métodos para cuantificar la aterosclerosis en conejo- •
Cuantificar las células espumosas (teñidas con Oil Red O) en la pared de la arteria.
- •
Extensión de lesiones con Sudan IV en aorta en face.
- •
Inmunohistoquímica (IH): permite cuantificar determinados marcadores. Así, por ejemplo, para cuantificar CML, realizaremos IH para detectar α-actina; si nuestro objetivo es los macrófagos, IH con RAM 11. Así, por ejemplo, nuestro grupo demostró que la inyección de apoA–I Milano a un modelo de aterosclerosis de conejo con doble denudación aórtica y dieta al 0,2% de colesterol facilitaba la regresión de la placa y la estabilizaba (disminuyó el porcentaje de células teñidas con RAM 11 y aumentó el de células teñidas con α-actina).
- •
Técnicas no invasivas de evaluación, como angiografía por tomografía computarizada (TC) y resonancia magnética (RM). Ampliamente utilizada por nuestro grupo17–29, permite evaluar el tamaño de placa de manera no invasiva. Se puede usar para estudios de progresión y regresión de placa, pues permite realizar varios estudios de manera seriada (es decir, monitorizar la respuesta con el paso del tiempo). De esta manera, cada animal es su propio control, lo que permite reducir de manera significativa el tamaño muestral.
Nuestro grupo usa una RM de 1,5 T y un coil convencional para extremidades. Se usan secuencias de ecografía de gradiente en cortes coronales y sagitales para localizar exactamente la aorta abdominal. A continuación, se realizan cortes secuenciales en el plano transversal (3mm de espesor, sin separación entre cortes) desde la salida del tronco celiaco a la bifurcación de las iliacas. Las imágenes inicial y final se identifican usando las distancias desde las arterias renales y la bifurcación iliaca, para obtener datos de cortes secuenciales. Se analizan los 5cm de la aorta inmediatamente tras el tronco celiaco. Mediante un programa de cuantificación de imágenes (Image J o Image Pro Plus), se cuantifican el área luminal y el área total; área de pared vascular = área total vascular – área luminal. Volumen de placa = área de pared vascular × 3 (pues los cortes son de 3mm de espesor). Se analiza la media de las medidas. Las figuras 1 y 2 muestran ejemplos del modelo de aterosclerosis en conejo.
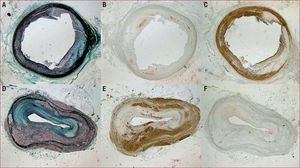
 y con placebo (D-F) en un modelo de conejo de dieta rica en colesterol y doble denudación aórtica. A y D: tinción con tricrómico de Masson; obsérvese que en A la estenosis es mucho menor que en D y con mayor proporción de colágeno (color púrpura) respecto al total de la placa que en D. B y E: inmunohistoquímica con RAM 11 (marcador de macrófagos); obsérvese que en A apenas hay macrófagos en la sección, mientras en E la infiltración de macrófagos es muy grande. C y F: inmunohistoquímica con actina (marca CML); obsérvese que la proporción de CML en C es mucho mayor que en F. En conclusión, la primera placa (A-C, tratada con apoA-I Milano) es mucho más estable (menos estenosis e infiltración macrofágica, mayor proporción de CML y de colágeno) que la segunda placa (D-E, placebo, más estenosis e infiltración macrofágica, menor proporción de colágeno y CML).")
Ejemplos de lesiones tratadas con apoA-I Milano (A-C) y con placebo (D-F) en un modelo de conejo de dieta rica en colesterol y doble denudación aórtica. A y D: tinción con tricrómico de Masson; obsérvese que en A la estenosis es mucho menor que en D y con mayor proporción de colágeno (color púrpura) respecto al total de la placa que en D. B y E: inmunohistoquímica con RAM 11 (marcador de macrófagos); obsérvese que en A apenas hay macrófagos en la sección, mientras en E la infiltración de macrófagos es muy grande. C y F: inmunohistoquímica con actina (marca CML); obsérvese que la proporción de CML en C es mucho mayor que en F. En conclusión, la primera placa (A-C, tratada con apoA-I Milano) es mucho más estable (menos estenosis e infiltración macrofágica, mayor proporción de CML y de colágeno) que la segunda placa (D-E, placebo, más estenosis e infiltración macrofágica, menor proporción de colágeno y CML).
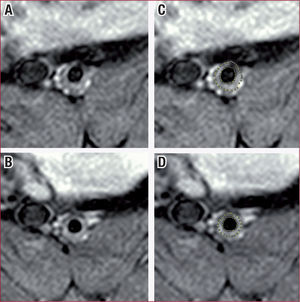
 de la regresión de la placa aterosclerótica en un conejo tras inyección de apoA-I Milano. Imágenes de RM antes (A) y después (B) del tratamiento (tomadas al mismo nivel de la aorta abdominal). C y D: las mismas imágenes que A y B, pero resaltando el autotrazado de la luz y el área total del vaso. Las mediciones por RM nos permiten cuantificar que en este segmento el tratamiento ha producido una regresión del 20,44% de la placa tras dos dosis de apoA-I Milano. Imagen obtenida de Ibanez et al27, con permiso del propietario del copyright.")
Evaluación mediante resonancia magnética (RM) de la regresión de la placa aterosclerótica en un conejo tras inyección de apoA-I Milano. Imágenes de RM antes (A) y después (B) del tratamiento (tomadas al mismo nivel de la aorta abdominal). C y D: las mismas imágenes que A y B, pero resaltando el autotrazado de la luz y el área total del vaso. Las mediciones por RM nos permiten cuantificar que en este segmento el tratamiento ha producido una regresión del 20,44% de la placa tras dos dosis de apoA-I Milano. Imagen obtenida de Ibanez et al27, con permiso del propietario del copyright.
Gottlieb fue el primero en comprobar en cerdo la presencia de aterosclerosis con engrosamiento intimal en arterias coronarias39. Genéticamente, el cerdo está relativamente próximo al humano, lo cual también se aplica a la anatomía y la fisiología del sistema cardiovascular y la respuesta a la hipercolesterolemia.
El cerdo doméstico (Sus scrufa) es el más usado en investigación cardiovascular. Alcanza la madurez sexual a los 6–8 meses, cuando su peso varía entre 40 y 100kg. El corazón del cerdo es anatómicamente similar al humano, con la excepción de que el ostium de la coronaria derecha está más craneal, que la vena ácigos izquierda permite el drenaje del sistema intercostal en el seno coronario, que es algo más proclive al vasospasmo y que no tiene colaterales40.
Ventajas: las lesiones son muy similares a las humanas, con acumulación de células espumosas, grasa extracelular, proliferación de CML, etc.; el perfil lipídico del cerdo es muy similar al humano, con la mayor parte del colesterol circulando en LDL y lipoproteínas en concentraciones y tamaños muy similares a las humanas; la única diferencia es que el cerdo no expresa apolipoproteína II (que supone el 20% del componente en apolipoproteínas del HDL); la anatomía cardiovascular es muy similar a la humana; la distribución de las lesiones es muy similar a la humana y predominan en aorta, coronarias y carótidas; las lesiones se desarrollan con dieta hipercolesterolémica; en general, se desarrollan estrías grasas a los 6 meses de edad y las lesiones avanzadas, en cerdos mayores de 1 año, con la única salvedad de que no se desarrolla trombosis; si se realiza la combinación de doble denudación aórtica y dieta hipercolesterolémica, las lesiones son aún más similares a las humanas41; los cerdos son omnívoros.
Inconvenientes: elevado gasto, dificultad de manejo y de almacenamiento; inducción aterosclerótica en tiempo relativamente largo; el cerdo habitualmente tiene concentraciones de cLDL relativamente bajas, por lo que no aparecen lesiones espontáneamente (colesterol total, 100 ± 8mg/dl; cLDL, 55 ± 8mg/dl; cHDL, 36 ±6 mg/dl; triglicéridos, 49 ± 4mg/dl)42; no obstante, cuando se lo alimenta con dieta rica en colesterol, se desarrollan hipercolesterolemia (300mg/dl aproximadamente) y lesiones ateroscleróticas similares a las humanas. No obstante, los tiempos de inducción son largos (9–12 meses) y las complicaciones de la placa (infarto, trombo), aunque descritas en la literatura, no son muy frecuentes. La tabla 4 muestra diversos modelos de inducción de aterosclerosis en el cerdo. Por ello, mediante selección, reproducción y cría adecuadas, se han desarrollado diversos modelos muy sensibles a la manipulación dietética con tiempos de inducción más cortos.
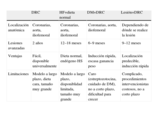
Métodos para desarrollar aterosclerosis en el cerdo.
| DRC | HF+dieta normal | DM+DRC | Lesión+DRC | |
| Localización anatómica | Coronarias, aorta, iliofemoral | Coronarias, aorta, iliofemoral | Coronarias, aorta, iliofemoral | Dependiendo de dónde se realice la lesión |
| Lesiones avanzadas | 2 años | 12–18 meses | 6–9 meses | 9–12 meses |
| Ventajas | Fácil, disponible universalmente | Dieta normal, endógeno HS | Inducción rápida, escasa ganancia peso | Localización predecible, inducción rápida |
| Limitaciones | Modelo a largo plazo, dieta cara, tamaño muy grande | Modelo a largo plazo, disponiblidad limitada, tamaño muy grande | Caro (estreptozotocina, cuidado de DM), no a corto plazo, dificultad para crecer | Complicado, procedimientos intervencionistas costosos, no a corto plazo |
DM: diabetes mellitus; DRC: dieta rica en colesterol; HF: hipercolesterolemia familiar.
Se ha desarrollado otra raza especial, inherited hyperlipoproteinemia and hipercolesterolemia (IHLC), en la que aparecen espontáneamente (con dieta normal) hipercolesterolemia, tasa de catabolismo de LDL reducida y aterosclerosis acelerada (incluso con hemorragia en la placa)43,44 debido a mutaciones en LDL-R.
A los 12 meses, los estadios iniciales de la lesión ya aparecen (macrófagos cargados de lípidos); las lesiones progresan rápidamente en 12–18 meses (principalmente en aorta torácica y bifurcación aortoiliaca); a las 24 meses hay lesiones complicadas (capa fibrosa fina, núcleo necrótico, calcificación, neovascularización) en casi todas las arterias, incluidas las coronarias; se ha descrito a los 36 meses presencia de muy abundante neovascularización, hemorragia en la placa y rotura de placa.
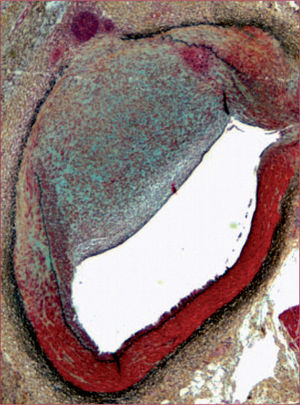
Las lesiones desarrolladas son similares a las humanas (estenosis, núcleo necrótico, neovascularización marcada), pero su principal inconveniente es que el tiempo de inducción es largo (12–18 meses, con el problema de manejo de cerdos que pueden llegar a superar 200kg de peso). La figura 3 muestra ejemplos de lesiones ateroscleróticas en cerdo.
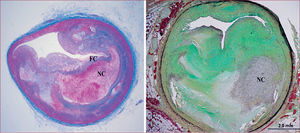
40. A la izquierda, placa con núcleo necrótico (NC) y recubrimiento fibroso fino (FC); el colágeno se ve en azul (tricrómico de Masson). A la derecha, placa complicada que contiene colágeno (en verde, pentacrómico de Movat), calcificación, lípidos, inflamación y núcleo necrótico (NC).")
Lesiones similares a las humanas en el modelo de cerdo con hipercolesterolemia familiar (IHLC)40. A la izquierda, placa con núcleo necrótico (NC) y recubrimiento fibroso fino (FC); el colágeno se ve en azul (tricrómico de Masson). A la derecha, placa complicada que contiene colágeno (en verde, pentacrómico de Movat), calcificación, lípidos, inflamación y núcleo necrótico (NC).
La combinación de diabetes mellitus (DM) e hipercolesterolemia en el cerdo Yorkshire induce lesiones ateroscleróticas en aorta, coronarias y arterias femorales en unas 20 semanas45,46. La DM se induce mediante administración intravenosa de estreptozotocina, que destruye más del 80% de las células beta pancreáticas). La dieta rica en colesterol acelera la aterogénesis (de hecho, en los cerdos diabéticos con dieta normal no se desarrollan lesiones).
A los 3 meses ya aparecen estrías grasas; a los 6 meses, el 96% de las arterias tienen lesiones, y el 100% a los 9 meses (el 19% con fibroateroma y el 11% de calcificación). En este modelo predominan las lesiones en las coronarias, iliacas y otras arterias periféricas, y son menos frecuentes en carótidas y aorta; en las coronarias, las lesiones más graves tienen una distribución similar que en humanos, en el territorio proximal y cerca de bifurcaciones. En este modelo se testó el efecto de darapladib, un inhibidor selectivo de Lp-PLA2 del que se ha demostrdo que reduce el desarrollo de aterosclerosis y núcleo necrótico47.
Una ventaja de este modelo es que los cerdos diabéticos ganan peso más lentamente que los cerdos normales o hipercolesterolémicos. Los inconvenientes residen en el precio y las complicaciones médicas de la DM que pueden sufrir los cerdos si no se monitorizan adecuadamente (hipoglucemia e hiperglucemia, gastroparesia, infecciones) (fig. 4).
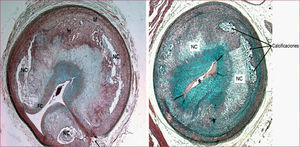
, núcleo necrótico (NC) y fino recubrimiento fibroso (NC). A la derecha, placa complicada con estenosis muy avanzada, núcleo necrótico (NC) y calcificaciones.")
Lesiones avanzadas en un modelo de cerdo diabético e hipercolesterolémico40. A la izquierda, placa aterosclerótica compleja, que contiene hemorragia (H), núcleo necrótico (NC) y fino recubrimiento fibroso (NC). A la derecha, placa complicada con estenosis muy avanzada, núcleo necrótico (NC) y calcificaciones.
El problema del tamaño se puede superar parcialmente empleando minicerdos, más económicos y con alta susceptibilidad a sufrir aterosclerosis inducida por dieta. La raza más usada es el cerdo en miniatura de raza Yucatán48–51; asimismo se han descrito modelos con las razas Göttingen, Hanford y Sinclair Hornell, pero la información es mucho menor. Se suelen requerir dietas muy ricas en colesterol para inducir aterosclerosis en minicerdos52 o inducción de DM con estreptozotocina. En el cerdo de Yucatán (pequeño y dócil) se desarrollan lesiones complicadas similares a las humanas, con necrosis, cristales de colesterol y calcificaciones. Muy recientemente se han comunicado avances que pueden cambiar sustancialmente el panorama en este campo (véase «Adenda»).
Modelo de lesión vascular + dieta hipercolesterolémicaPara disminuir el tiempo de inducción de las lesiones y reducir el coste del mantenimiento del cerdo, se han desarrollado modelos de aterogénesis mediante la combinación de dieta rica en colesterol y producción de lesión vascular localizada (denudación endotelial mediante guía53, inflado de balón endovascular50, ligamiento parcial de la carótida54).
Una técnica útil descrita recientemente es la inyección de solución lipídica directamente en la pared de los vasos del cerdo hipercolesterolémico55,56, lo cual crea a los 12 meses lesiones excéntricas, con remodelado positivo, cubierta fibrosa fina, núcleo necrótico y abundante neovascularización. Se alimenta al cerdo con dieta rica en colesterol (el 2% de colesterol, el 20% de grasa, el 1,5% de colato sódico), se lo somete a un inflado de balón de angioplastia y, 2 semanas después, se inyecta una mezcla de liposomas que contienen ésteres de colesterol y LDL humana oxidada.
La principal ventaja del modelo es que la distribución anatómica de las lesiones es predecible, únicamente se desarrollarán donde hayamos producido la lesión vascular (que será selectivamente en el territorio que más interese). El principal inconveniente es la complejidad de inducir la lesión vascular (fig. 5, tabla 4).

Placa en la arteria descendente anterior de un cerdo sometido a inyección de lípidos en la pared vascular 10 semanas antes. En esta tinción con el pentacrómico de Movat, se observa una placa fibrolipídica56.
Dado que es el animal más similar al ser humano en la escala filogenética, es un modelo muy atractivo, pues se podrá aplicar los hallazgos más directamente al ser humano. Los primates americanos en general no se usan porque se desarrollan lesiones discordantes, con distribución anatómica diferente de la del hombre. Los más usados son los primates africanos porque tras dieta hipercolesterolémica se desarrollan lesiones en localizaciones anatómicas muy similares a las humanas. Por ejemplo, el mono Rhesus es el más estudiado, con las ventajas de un tamaño adecuado y unas lesiones ya bien caracterizadas. Similares ventajas comparten los monos Cynomolgus.
Ya en los años cincuenta, se consiguió inducción de aterosclerosis en monos con dieta rica en colesterol57, incluso con aparición de IAM58. Dicha dieta puede inducir lesiones en la aorta y todas sus ramas, incluidas las arterias coronarias. Recientemente se ha descrito un modelo de mono Rhesus con deficiencia familiar de LDL-R59,60.
Ventajas: filogenéticamente similares a los humanos; son omnívoros; las lesiones son similares a las humanas, con alto porcentaje de proliferación de músculo liso y complicaciones (trombos, infartos, etc.).
Inconvenientes: elevado precio, mantenimiento caro, escasa disponibilidad, animalarios especiales, problemas éticos.
AdendaEn el lapso entre la redacción de este artículo y la revisión de las pruebas, se ha publicado por primera vez la creación de un modelo de cerdo Yucatán modificado genéticamente para expresar la proteína mutante humana PCSK9 (causa de la hipercolesterolemia familiar). Este cerdo enano desarrolla espontáneamente hipercolesterolemia, y cuando se lo alimenta con dieta grasa, sufre un proceso aterosclerótico muy similar al de los humanos70.
Conflicto de InteresesNinguno.