




Palabras clave
INTRODUCCIÓN
A pesar de los grandes avances que se han hecho en cardiología, las enfermedades cardiovasculares siguen siendo la principal causa de muerte y morbilidad en las sociedades industriales. El siglo xx se ha caracterizado por el rápido desarrollo de la tecnología médica, por la rápida expansión de la información relacionada con la investigación y el desarrollo ideológico, y por la relativamente rápida incorporación de información nueva a la práctica. Los avances en los métodos de diagnóstico y terapia en la cardiología clínica han conducido a un progreso real en el tratamiento y, en algunos casos, a una mejoría o incluso prevención de las enfermedades cardiovasculares. Sin embargo, a pesar de la progresiva sofisticación de las técnicas, nuestra capacidad para tratar las causas de las enfermedades cardiovasculares sigue siendo limitada. En otras palabras, podemos tratar muchos de los síntomas, pero debido a la limitación en la comprensión de la etiología primaria que subyace al comienzo o desarrollo de un proceso patológico, las causas desencadenantes siguen sin poderse modificar por la práctica clínica.
Hasta muy recientemente, los enfoques que se han utilizado para estudiar las enfermedades se han basado en herramientas farmacológicas, quirúrgicas, bioquímicas o fisiológicas. Constituye una lección de humildad el darnos cuenta de que estamos tratando de describir sistemas fisiológicos complejos sin tener una comprensión o una apreciación completa de las identidades de las partes integrantes. Las estimaciones sobre el número de genes en el genoma de los mamíferos varían desde 50.000 a 100.000, pero hasta el momento sólo el 10% parecen tener algún tipo de utilidad. Así pues, es posible que estemos perdiendo aproximadamente un 90% de la información más básica necesaria para entender el rendimiento genético una información que casi con seguridad está en la base de muchas de las enfermedades cardiovasculares, de manera directa o indirecta, al determinar la respuesta del corazón a los estímulos ambientales. Mientras se escribe este trabajo de revisión, diversos comunicados han informado sobre la finalización del proyecto genoma humano. Durante los próximos 1-3 años podremos comparar esta información con la obtenida sobre el genoma de otros mamíferos, tales como la rata y el ratón. Aunque estos datos aportan las bases estructurales para una comprensión profunda de la fisiología de los mamíferos, tomados tal y como se obtienen pueden ser de muy poca ayuda para el clínico. Su valor va a depender en gran medida de la eficiencia con la que los científicos básicos sean capaces de extraer información de estas bases de datos. La utilización de los datos que se obtienen en su contexto biológico puede conducir a un nuevo concepto de medicina. En su conjunto, todos estos datos pueden, por una parte, revelar la etiología genética primaria responsable del desarrollo de las enfermedades y, por otra, proporcionar nuevas estrategias de manejo en el tratamiento de las enfermedades cardiovasculares.
Si en los últimos 25 años la biología molecular nos ha enseñado algo acerca de la biología clínica es, precisamente, que los elementos proteicos específicos subyacen a la función de las células y de los órganos. A su vez, la producción proteica está controlada por patrones de transcripción específicos para cada tipo celular y definidos por un conjunto común de genes. Puesto que la célula y, en último término, la función de un órgano depende de los polipéptidos que están presentes, no es sorprendente que cuando la función está alterada, por ejemplo durante el desarrollo de una enfermedad, se produzcan también cambios en el contenido proteico. En el corazón existen numerosos ejemplos de cambios en las proteínas contráctiles que se correlacionan con alteraciones funcionales, tanto durante el desarrollo normal como durante el proceso de desarrollo de numerosas patologías. De igual manera, varias enfermedades congénitas cardíacas se caracterizan por determinados cambios en las proteínas motoras. Así por ejemplo, en la insuficiencia cardíaca se han documentado cambios en la cascada de regulación de diversos (hipotéticos) efectores/moduladores. Para entender todas estas relaciones y poder establecer modelos en los cuales los procesos patogénicos puedan ser estudiados de forma longitudinal, es necesario dirigir el corazón para que sintetice de forma estable, en ausencia de otros cambios pleiotrópicos, la proteína candidata. De esta forma es posible determinar si la presencia de una proteína es responsable directa o indirectamente de los efectos estudiados, con el objetivo de definir las potenciales dianas terapéuticas. Al manipular el contenido proteico del corazón de una determinada manera, se dispone de un medio adecuado para establecer tanto el mecanismo como la función de diferentes proteínas mutadas o isoformas proteicas. La transgénesis y la reconstrucción génica, y la subsiguiente creación de modelos animales estables, proporcionan herramientas para modificar el genoma de los mamíferos y estudiar las consecuencias que esto pueda tener en el sistema cardiovascular. Utilizando la expresión dirigida al corazón de una determinada proteína modificada por ingeniería genética, se puede remodelar de manera efectiva el perfil de proteínas cardíacas y también estudiar las consecuencias de una manipulación genética aislada a nivel molecular, bioquímico, citológico y fisiológico, tanto en condiciones normales como bajo estímulos de estrés.
Nuestra discusión se centrará en las miocardiopatías, y en cómo los modelos obtenidos por ingeniería genética han sido utilizados de manera eficaz para confirmar la etiología primaria y para explorar los procesos patogénicos que finalmente conducen a una insuficiencia cardíaca. Se analizarán las ventajas y los puntos débiles de la tecnología que se encuentra actualmente disponible, así como la que está en proceso de desarrollo, y que tiene un futuro prometedor en este campo. En lugar de ser una revisión de tipo enciclopédico, este trabajo se centrará en la manera en cómo los conceptos actuales se están aplicando a su objetivo, para que el desarrollo de futuras tecnologías, que in-
evitablemente nos van a sorprender, puedan ser apreciadas en su contexto.
CREACIÓN DE MODELOS ANIMALES ESTABLES
Existen varias técnicas disponibles hoy día que permiten alterar el aparato contráctil del corazón de una manera determinada: nos vamos a centrar en aquellos enfoques que resultan en una transmisión por la línea germinal de los eventos de remodelado. De esta manera, las modificaciones deseadas pueden ser propagadas a través de sucesivas generaciones y pueden resultar en la creación de modelos animales nuevos y estables. Necesariamente estos cambios estables tienen que realizarse a nivel genético y con este objetivo se han desarrollado 2 aproximaciones distintas pero complementarias: la transgénesis y la reconstrucción génica. Cada una de ellas sirve para modificar el genoma de los mamíferos pero son muy diferentes entre sí, tanto en los aspectos técnicos como en su rendimiento genético. Mediante la transgénesis, el aparato contráctil del corazón puede ser remodelado de manera significativa, pero los resultados del método residen exclusivamente en la capacidad de los transgenes para producir un fenotipo que se contraponga al contexto endógeno, ya que los transgenes no afectan el rendimiento genético endógeno. De igual manera que la transgénesis, la reconstrucción génica (a menudo referida como ablación génica o eliminación génica) cambia de forma directa la estructura genética, y el aparato contráctil del corazón puede ser alterado drásticamente con esta técnica. La eliminación de un gen puede conducir a la pérdida de una función, lo que permite establecer una secuencia posible responsable de esta función. La reconstrucción génica también puede usarse para producir cambios en las secuencias que codifican un dominio funcional de una proteína contráctil, o cambios a nivel de un único aminoácido, lo que permite establecer relaciones precisas entre estructura y función. Todas estas aproximaciones experimentales, que se encuentran comparadas y contrastadas en la tabla 1, están dando lugar a un grupo de animales en los que las alteraciones en los elementos proteicos contráctiles conducen a una comprensión profunda de las relaciones entre estructura y función que subyacen a la función cardíaca a nivel molecular, bioquímico, orgánico y también a nivel del animal entero.

Transgénesis
La transgénesis permite alterar de manera estable el genoma de los mamíferos de tal manera que las modificaciones puedan ser transmitidas a través de la línea germinal. Aunque el procedimiento es técnicamente posible en los animales grandes1, debido al coste económico, al tiempo y a consideraciones en el manejo de los animales, la investigación básica se ha limitado fundamentalmente al ratón y, en casos excepcionales, al conejo2,3. El desarrollo de esta técnica, que consiste en la inyección de ADN directamente en el embrión fertilizado, procede del desarrollo de técnicas relacionadas con los campos de la manipulación embrionaria y el ADN recombinante4. La inserción de ADN exógeno es un proceso que se produce al azar y numerosas copias se insertan dentro del genoma, normalmente en una disposición paralela o antiparalela. El investigador no puede controlar el punto de inserción, ni tampoco predeterminar el número de copias que se colocan en el genoma receptor. Las diversas fases de este proceso se describen en la tabla 1. Así, a diferencia de la reconstrucción génica, en la cual la recombinación homóloga puede ser seleccionada (ver más adelante, en la siguiente sección), la transgénesis es un fenómeno que se produce al azar, y el ADN se sitúa en lugares distintos a los que se encuentran las secuencias homólogas. Esto conlleva que la expresión de los transgenes se superimponga a la expresión de los genes endógenos. Así, para que se manifieste un fenotipo, el transgén tiene que producir un efecto dominante. La actividad transcripcional final del ADN transgénico puede ser dependiente o independiente del número de copias, y a menudo se encuentra influenciada por la localización cromosómica particular en la cual está insertado («efecto posicional»), haciendo que sea más o menos activo desde el punto de vista transcripcional. Además, la inserción del ADN no es un proceso benigno y puede producir una importante disrupción, reorganización o pérdida del ADN que lo flanquea, lo que da lugar a efectos mutagénicos por inserción5. Afortunadamente estos efectos son mayoritariamente recesivos y, dado que los análisis de los animales transgénicos suelen estar restringidos a los heterocigotos, esto no representa un problema. Los efectos debidos al número de copias y a la posición requieren el análisis de múltiples lí neas, lo que constituye un proceso costoso e intensivo. No obstante, se pueden generar múltiples mutaciones y constructos quiméricos y se han conseguido numerosas líneas de ratones de forma relativamente rápida si se compara con la técnica de reconstrucción génica. Por todos estos aspectos se recomienda una consideración cuidadosa sobre el tipo de enfoque transgénico para los estudios de remodelado cardíaco.
La expresión sistémica de un transgén puede complicar seriamente cualquier fenotipo cardíaco resultante. El desarrollo de promotores o secuencias que controlan y dirigen la expresión de los genes, y cuya expresión está fundamentalmente restringida al corazón, ha mejorado la utilidad del enfoque transgénico para el estudio de las enfermedades cardiovasculares en ausencia de efectos en otros órganos o tejidos musculares que puedan inducir a la confusión. No es sorprendente que las propias secuencias transcripcionales de los genes del aparato contráctil cardíaco hayan sido usadas para dirigir de manera efectiva la expresión transgénica cardíaca. Entre estas secuencias se encuentran los promotores de la actina, de la cadena ligera de la miosina 2v, y de las cadenas pesadas de la miosina * y ß, entre otras6. Se han expresado un gran número de transgenes (cientos de ellos) que afectan al corazón utilizando los promotores cardíacos específicos y se han creado modelos en los que se producen eventos de remodelado general, tales como la hipertrofia, o enfermedades cardíacas concretas7. Es interesante señalar que los resultados, en términos de niveles proteicos totales, pueden variar ampliamente en los diferentes experimentos transgénicos, dependiendo de la proteína que se intenta crear. Así por ejemplo, tal como veremos más adelante, muchas de las miocardiopatías hipertróficas están causadas por mutaciones en las proteínas contráctiles8. Se ha llevado a cabo un estudio detallado del paradigma transgénico utilizando muchas de las otras proteínas contráctiles. Estos estudios muestran que, a pesar de haber una sobreexpresión importante a nivel del ARN, los niveles proteicos permanecen inalterados. Puesto que el ARN transgénico se produce en mucha mayor cantidad que el ARN endógeno, el efecto neto en el cardiomiocito es el de diluir la proteína endógena con cantidades crecientes del polipéptido transgénico. El resultado final es que se produce un reemplazo de la proteína endógena por la que es codificada de manera transgénica9.
Es importante entender las limitaciones que tiene la aproximación experimental transgénica, muchas de las cuales ya han sido mencionadas anteriormente. En primer lugar, conviene insistir en ello, esta técnica se basa en el hecho de que cualquier transgén que se exprese tiene que «dominar» el producto génico endógeno. En segundo lugar, hay que tener presente la posibilidad de una potencial mutagénesis por inserción, incluso en el estado heterocigótico. En tercer lugar, no hay que olvidar la cuestión de la sobreexpresión. Como se ha dicho anteriormente, en el caso de las proteínas contráctiles este aspecto no parece ser un problema. Además, como grupo control, la proteína original puede ser expresada a un nivel similar para asegurarnos de que el fenotipo no está presente. Sin embargo, los promotores que se utilizan normalmente en estos experimentos proceden de las proteínas contráctiles. Estos polipéptidos constituyen la mayor parte de la masa proteica de los cardiomiocitos y se expresan en gran cantidad, con un estado de equilibrio en el número de transcriptos de ARN que varía de diez a miles. Así pues, un promotor muy activo es parte del constructo transgénico. En muchos experimentos en los que se expresan transgénicamente sustancias potencialmente tóxicas, potentes moléculas de señalización biológica o moléculas amplificadoras, los resultados son muy difíciles de interpretar. Por ejemplo, en una publicación reciente se expresó el gen marcador Green Fluorescent Protein (GFP) usando un promotor específico cardíaco10. La hipertrofia resultante fue interpretada como que la sobreexpresión de cualquier molécula puede resultar en hipertrofia. Si examinamos la bibliografía general, veremos claramente que esto no es cierto. Otra explicación posible es que la expresión de GFP en el contexto del corazón comprometió la función y dio lugar a la hipertrofia resultante. Es decir, la GFP no es una proteína dañina, sino que es capaz de producir una respuesta hipertrófica. Así, en un experimento en el que se investiga la ganancia de una función, y en el que no hay un grupo control que exprese la proteína original de forma paralela a la proteína mutante que se está estudiando, la interpretación de los resultados puede ser difícil o imposible.
Reconstrucción génica
La reconstrucción génica (en inglés gene targeting), a diferencia de la transgénesis, reemplaza la secuencia de ADN endógeno, en un punto previamente seleccionado, con ADN preparado exógenamente (y, es de suponer, que alterado). La reconstrucción génica utiliza la recombinación homóloga para insertar el ADN en lugares específicos de los genes, lo que produce una inactivación (o mutación) del gen de interés, y genera animales deficientes (o que tienen una mutación) en ese producto génico concreto. Esta tecnología se usa típicamente para inactivar o conseguir genes «knock outs» en el genoma y de ahí que reciba el nombre de experimento de «pérdida de función». Desde el punto de vista técnico es un procedimiento muy diferente a la transgénesis y mucho más largo. Los enfoques convencionales de reconstrucción génica se basan en la recombinación homóloga de ADN electroporado dentro de células madre embrionarias totipotentes11. El ADN electroporado contiene normalmente un marcador que sirve para identificarlo, de manera que después de los correspondientes procedimientos de selección, aquellas células en las que ha tenido lugar una recombinación homóloga pueden ser identificadas mediante procedimientos de cribaje molecular, tales como el análisis por Southern o PCR12. Existen muchas variaciones sobre este procedimiento básico, pero los aspectos principales permanecen más o menos invariables. El ADN que se inserta consiste en regiones extensas de homología, que flanquean el lugar de interés y que contienen al menos un marcador, y a veces dos marcadores. El proceso de reconstrucción se planifica para que se produzca una eliminación de las secuencias endógenas con elementos de ADN críticos que hacen que la transcripción/traducción del gen pueda ser controlada y reemplazada por un marcador seleccionado o por una secuencia inocua no codificadora. Esto da lugar a un «alelo nulo», es decir, un locus génico que no puede producir un producto de transcripción que pueda ser traducido y/o un polipéptido funcional. La ablación génica o reconstrucción permite estudiar las consecuencias de las alteraciones recesivas si los animales resultantes son cruzados hasta conseguir homocigotos.
Esta estrategia es utilizada fundamentalmente para determinar si un gen concreto tiene una función crítica, o para explorar la potencia del efecto del gen dependiente del número de copias mediante la elimi nación de uno de los alelos. Si el proceso de reconstrucción da lugar a un transcripto alterado pero que puede ser traducido, se puede formar un «péptido tóxico» que interfiere con la función de la proteína endógena, lo que se conoce como una reconstrucción de tipo «negativo dominante». Además, la recombinación homóloga en células madre embrionarias se puede usar para insertar, en un punto definido, un gran fragmento de ADN que lleve incorporado un transgén entero único13, de tal manera que su actividad y función (o funciones) puedan ser estudiadas sin que haya interferencias con los efectos debidos al número de copias o a los diferentes contextos cromosómicos.
En la bibliografía existen numerosos ejemplos de genes que han sufrido una ablación y que son importantes para el desarrollo general y la función del corazón. A menudo, estos genes remodelados codifican polipéptidos que están pleiotrópicamente involucrados en el desarrollo cardíaco o la estructura básica. Mientras que la ablación puede tener un impacto importante sobre el aparato contráctil, la deficiencia/remodelado no es una consecuencia primaria de la mutación nula sino que refleja más bien un déficit más básico que ha tenido lugar en el desarrollo cardíaco normal. Ejemplos de ello lo constituyen el «knockout» de hox-1.5, que da lugar a la generalización de anormalidades cardiovasculares reminiscentes del síndrome de DiGeorge14, o la ablación de la proteína LIM específica del músculo15 que produce una miocardiopatía dilatada, así como los «knockouts» de endotelina-116 y conexina 4317. La eliminación de estos genes, cuyas funciones son aparentemente necesarias para el desarrollo cardíaco normal y para el establecimiento de la estructura inherente al miocardio, ha tenido un gran valor informativo, pero no ha aportado una evidencia directa que mejore la comprensión de las relaciones que existen entre estructura y función y que subyacen a la función cardíaca normal y anormal en el adulto.
Las ventajas de la reconstrucción génica sobre la transgénesis pueden ser sustanciales. En primer lugar, se conoce tanto el punto de inserción como el número de secuencias insertadas. En segundo lugar, el ADN mutado se inserta sólo en un punto definido. En tercer lugar, si se produce una mutación sin que haya ablación, esa mutación se expresará bajo el control del promotor endógeno, lo que produce niveles de expresión fisiológicamente relevantes, que mimetizan exactamente los patrones de transcripción endógenos del gen que se está estudiando. Sin embargo, por diversas razones, los resultados relacionados directamente con el remodelado del corazón a través de la ablación o la modificación de genes, especialmente aquellos que codifican elementos del aparato contráctil, son limitados.
Es de esperar que la ablación de un componente crítico del aparato contráctil sea letal. Un corazón funcional es crítico para el desarrollo fetal en el día 11-12 del período embrionario del ratón. Si el corazón se encuentra seriamente comprometido, el feto raramente sobrevive más allá del día 12,5 del período embrionario. Así pues, los animales homocigotos que contengan una mutación nula en los 2 alelos no llegarían a término, morirían durante la embriogénesis y serían reabsorbidos18. Antes de entrar en las dificultades de crear un animal genéticamente reconstruido es recomendable tener la infraestructura analítica necesaria, que no ha estado disponible durante mucho tiempo en el caso de la evaluación de la función cardíaca en el útero. Nuestra capacidad para discernir una función contráctil cardíaca en el embrión intacto a media gestación, medida por el volumen de eyección y la frecuencia cardíaca, ha sido bastante limitada hasta ahora pero está mejorando rápidamente19. Otro aspecto crítico es que la reconstrucción génica es una técnica que requiere mucho tiempo para llevarse a cabo, y no es infrecuente que un experimento dure años (mientras que puede tardar meses si se utiliza el enfoque transgénico). Esto es especialmente cierto cuando se trata de provocar una mutación en el gen, y no simplemente eliminarlo, ya que este tipo de procedimientos requiere 2 procesos de reconstrucción separados20. No obstante, la reconstrucción génica sigue siendo, entre las 2 posibilidades, la aproximación experimental más elegante e intelectualmente satisfactoria, por ser la más precisa.
MODELOS OBTENIDOS POR INGENIERÍA GENÉTICA DE ENFERMEDADES CARDIOVASCULARES
Miocardiopatía hipertrófica familiar
Las miocardiopatías hipertróficas familiares (MHF) incluyen un grupo de desórdenes musculares cardíacos primarios caracterizados por una gran morbilidad y mortalidad. Su clasificación está basada en su fisiopatología y etiología. La miocardiopatía se definió originariamente como una enfermedad no coronaria del músculo cardíaco21. En 1980 la Organización Mundial de la Salud redefinió el término como una enfermedad cardíaca de etiología indefinida. Aunque se conocen varias miocardiopatías de diferentes categorías (dilatada, hipertrófica, restrictiva/obliterativa), nuestra discusión se centrará fundamentalmente en las MHF. Este tipo de miocardiopatías son heredadas, más que adquiridas como consecuencia de una carga cardíaca alterada22, son autosómicas dominantes y dan lugar a una miocardiopatía hipertrófica medida como un aumento de la masa. Las MHF se caracterizan por una hipertrofia cardíaca inexplicable sin que haya un aumento de la carga cardíaca, o en ausencia de otras anormalidades sistémicas. Desde el punto de vista clínico, las enfermedades autosómicas dominantes tienen una penetrancia variable con ocurrencia de hipertrofia en cualquier ventrículo; normalmente (en > 95%) también está involucrado el septum intraventricular23. Histológicamente se observa una desorganización característica de los miocitos y de las miofibrillas. A menudo también se puede apreciar una morfología nuclear extraña y cariomegalia, aunque estas características pueden ser dependientes de la edad y no estar presentes en los niños24. Los hallazgos histopatológicos también incluyen a menudo una fibrosis extensiva que puede ser focal o intersticial25. Como grupo de enfermedades, las MHF son bastante comunes, y hay estudios independientes que estiman que uno de cada 500 individuos está afectado26,27. Las MHF son una causa importante de muerte súbita en adultos jóvenes aparentemente sanos28. Un estudio reciente ha calculado que entre 35 adultos jóvenes que presentaron muerte cardíaca súbita, 12 fueron por causa de MHF29. Incluso dentro de una misma familia en la que la enfermedad es debida a un único defecto genético, la severidad, inicio y penetrancia de la patología es altamente variable (probablemente debido a la existencia de loci modificadores, aunque esto no se ha podido demostrar todavía de manera concluyente). La penetrancia es dependiente de la edad, con un comienzo que ocurre normalmen te en la adolescencia25,30,31.
El decisivo artículo de Geisterfer-Lowrance et al32 fue el primero en establecer una base genética en la etiología primaria de esta enfermedad. Mediante técnicas de ligamiento génico acopladas con aproximaciones experimentales de biología molecular, y aprovechando su gran experiencia, el grupo de Seidman estableció que los trastornos ligados a la FHC se encontraban localizados en el cromosoma 14q11. Ellos fueron los que demostraron que en todos los individuos afectados de la misma familia había una mutación puntual en el exón 13 del gen de la cadena pesada ß de la miosina cardíaca (ß-MyHC). Este gen codifica las principales proteínas motoras responsables del bombeo del ventrículo adulto. La mutación convertía un residuo de arginina altamente conservado (Arg403) en uno de glutamina (Arg403Gln). Desde esta observación inicial, se han mapeado más de 50 diferentes mutaciones puntuales de la ß-MyHC que producen varias sustituciones de aminoácidos: las mutaciones en este gen son responsables de aproximadamente un 30-40% de las MHF en la población general. Este gran número de mutaciones diferentes, y su localización variable dentro de la molécula de MyHC ayuda, en parte, a explicar la enorme variabilidad en la severidad de esta enfermedad: mutaciones diferentes tienen efectos diferentes sobre la capacidad de la MyHC para realizar su función. Por ejemplo, la mutación Arg403Gln es altamente penetrante y tiene efectos severos sobre la morbilidad y mortalidad de la población afectada. El residuo está localizado cerca del lugar de unión del nucleótido, y es en este punto donde la cabeza de la miosina participa en la interacción actina-miosina. Así pues, la mutación se encuentra en y cerca del lugar crítico para ejercer las funciones motoras33.
La genética de las miocardiopatías dilatadas y arritmogénicas no es tan bien conocida ni comprendida como la de las MHF, aunque se han descrito varios loci génicos y se han identificado unos pocos genes34,35. Igual que en el caso de la MHF, este tipo de desórdenes puede presentar características clínicas heterogéneas y una morbilidad variable, y otros genes modificadores ejercen, sin duda, influencia sobre la mortalidad.
Actualmente sabemos que la MHF es un desorden genético autosómico dominante que se deriva de mutaciones en diferentes genes codificadores de proteínas sarcoméricas que constituyen los filamentos gruesos, finos y los filamentos de titina. Se han identificado múltiples mutaciones en los genes de la ß-MyHC, las cadenas ligeras esencial y reguladora de la miosina (ELC y RLC), la proteína C que se une a la miosina (MyBP-C), la tropomiosina (*-Tm), la troponina T cardíaca (cTnT), la troponina I (cTnI), la actina y la titina8,36. Clínicamente la MHF induce eyección hiperdinámica, relajación reducida, retraso en el llenado inicial, desorganización de los miocitos y fibrosis, y aumento de la rigidez cameral telediastólica. El diagnóstico normalmente se basa en el examen físico y ecocardiográfico, aunque en los niños es particularmente difícil hacer un diagnóstico correcto porque puede ser que no tengan hipertrofia cardíaca hasta que alcanzan la edad adulta.
Aunque el conocimiento de la etiología de la enfermedad a nivel de su genética molecular está muy avanzado, siguen sin conocerse bien los procesos patogénicos responsables de su morbilidad y mortalidad. Esto se debe a que la descripción de estos procesos tiene que realizarse necesariamente a lo largo de períodos prolongados de tiempo, y en una población en la que el comportamiento y el entorno no están controlados. Con el fin de mitigar estas dificultades, se está realizando un gran esfuerzo para generar modelos animales relevantes en los que la progresión de la enfermedad pueda ser rigurosamente estudiada en condiciones controladas. El desarrollo de estos modelos animales ha arrojado luz sobre la patogenia de la MHF, permitiendo establecer vínculos entre las mutaciones genéticas y los déficit funcionales concretos. En teoría, una proteína sarcomérica mutante que presumiblemente causa el desarrollo de una enfermedad puede alterar la función sarcomérica al menos por 2 mecanismos diferentes:
Puede funcionar como un «péptido tóxico», ejerciendo un efecto negativo dominante. La proteína alterada es incorporada en el sarcómero, lo que conduce a cambios estructurales y al desarrollo de hipertrofia compensatoria.
Puede actuar como un «alelo nulo», con capacidad potencial de producir haplo-insuficiencia. La producción de cantidades insuficientes de proteína normal produce un desequilibrio en la estoiquiometría del sarcómero, que altera la estructura y la función sarcoméricas.
Más adelante comentaremos brevemente ejemplos relevantes que ilustran el potencial de los modelos animales para descubrir los déficit de las enfermedades primarias.
La cadena pesada de la miosina
La cadena pesada de la miosina (MyHC) es, por su masa, el principal componente de los filamentos gruesos del sarcómero y proporciona la función motora de la contracción cardíaca. La miosina es una proteína hexamérica que está formada por 2 cadenas pesadas (PM aproximado de 220 KDa) y 2 pares de cadenas ligeras no idénticas (PM aproximado de 18-27 KDa). Las cadenas pesadas están formadas por 2 dominios separados: una cabeza globular unida a una cola *-helicoidal por una región bisagra. Las cabezas, localizadas en el extremo aminoterminal de la molécula, contienen la región catalítica de la ATPasa, y un lugar de unión para la actina. Además, las cadenas ligeras están unidas a cada cabeza por la región de unión cabeza-cola, también llamada «cuello», «región bisagra» o «subfragmento-1» (S1). Las cadenas pesadas de la miosina determinan las diferentes funciones motoras de los diferentes tipos musculares y constituyen una familia de genes37, y en el corazón se expresan 2 isoformas, la * y la ß. Las isoformas * y ß están codificadas por los genes MYH6 y MYH7, respectivamente, y están organizadas en tándem agrupadas en el cromosoma 14q11.2-q13, con el gen MYH7 localizado 4 kilobases por encima del MYH6. El gen MYH7 está compuesto por 40 exones, y codifica una proteína de 1.935 residuos de aminoácidos (fig. 1). Las isoformas * y ß están organizadas como homodímeros V1 y V3, respectivamente. Bajo determinadas condiciones, puede estar presente una forma heterodimérica intermediaria llamada V2, que está constituida por los productos génicos * y ß. Los homodímeros V1 y V3 tienen diferentes actividades ATPasa intrínsecas, siendo V1 la más activa (alrededor de tres o cuatro veces más). Estas diferentes isoformas están presentes en cantidades variables, y existe una buena correlación entre la velocidad de contracción y la relación entre V1/V3. Así, el ventrículo humano contiene fundamentalmente V338,39, mientras que el ventrículo de ratón adulto, que alcanza una frecuencia de 500-800 latidos por minuto, contiene V1.
En la población humana existen múltiples mutaciones en la ß-MyHC de la MHF. Hasta la fecha se han identificado más de 63 mutaciones distintas. Estas mutaciones se encuentran distribuidas a lo largo de la molécula, pero la mayor parte están localizadas en la mitad aminoterminal, con grupos en la región de la cabeza globular, en el lugar de la unión del ATP o de la actina, en los lugares de unión de la cadena ligera33, en la zona de unión cabeza-cola, así como en unos pocos lugares de la región de la cola. Se han descrito mutaciones sin sentido y mutaciones con pérdida de sentido, y deleciones, y esta diversidad es responsable, al menos en parte, de la gran variabilidad en la presentación clínica de la MHF. De este modo, el clínico se enfrenta a enormes diferencias en términos de grado de hipertrofia, inicio y evolución de los síntomas de la enfermedad, ocurrencia de muerte cardíaca súbita y pronóstico del paciente (clasificado como benigno o maligno). Los efectos de las diferentes mutaciones sobre las funciones de la cadena pesada de la miosina se acompañan de pronósticos distintos. Las principales mutaciones clínicas asociadas con un pronóstico maligno incluyen la Arg403Gln32,40-42, Arg453Cys33,43,44, Gly716Arg33,45,46, Arg719Trp45,47,48 y, dependiendo del origen étnico de los pacientes, Val606Met42,44,49-51. La sustitución Arg403Gln tiene consecuencias dramáticas, y el 50% de los individuos afectados mueren a la edad de 40 años41,52.
Con el objetivo de estudiar sus consecuencias y utilizando la técnica de la reconstrucción génica, se ha generado en el ratón la mutación con pérdida de sentido Arg403Gln (R403Q)53. Este ratón ha sido analizado extensivamente a lo largo de su vida y ha proporcionado información valiosa relativa a la historia natural de esta enfermedad. En los humanos la mutación sólo está presente en la forma heterocigótica y, de acuerdo con esta observación, el ratón homocigótico muere a los pocos días de nacer54. Los individuos heterocigotos sedentarios sobrevivieron, lo que demuestra que la presencia de la mutación de la miosina Arg403Gln da lugar a una función cardíaca anómala pero compatible con la vida. Los heterocigotos presentaron la misma histopatología y fisiopatología55-58 que se había observado en la forma humana de MHF. La disfunción cardíaca precedió los cambios histopatológicos, y la des organización miocitaria, hipertrofia y fibrosis aumentaron con la edad53. Estos datos han proporcionado una evidencia definitiva de la base genética de la MHF, y han demostrado que la disfunción cardíaca es la respuesta principal a la mutación Arg403Gln de la miosina. Es interesante señalar que los heterocigotos machos jóvenes presentaron un fenotipo más severo que las hembras, tal como se comprobó por la presencia de anormalidades electrofisiológicas que estuvieron ligadas al sexo59,60. Los machos se encuentran afectados a una edad más temprana que las hembras, aunque se desconocen las causas de estas diferencias ligadas al sexo. La capacidad para hacer ejercicio está comprometida en los heterocigotos53. En el futuro, este modelo de ratón para la MHF va a permitirnos continuar el estudio del impacto que tiene el genotipo, la dieta y la actividad física sobre el fenotipo.
Se han evaluado las consecuencias funcionales de la mutación Arg403Gln a nivel molecular61: la actividad cíclica de la actina está acelerada, y ésta puede ser la causa de la mayor velocidad en el aumento de la presión observada in vivo55, así como de la mayor velocidad en el desarrollo de fuerza en tiras musculares62. Esta ganancia de la función es consistente con el fenotipo hemodinámico observado en los humanos, y probablemente estimula el desarrollo de una hipertrofia compensatoria que no es debida a una disminución en la capacidad de generar fuerza sino que, por el contrario, está relacionada con una demanda energética crónicamente aumentada en el miocardio. En este modelo, la hipertrofia compensatoria aumentaría la masa tisular para reducir el estrés de pared y la utilización energética por unidad de volumen del miocardio. En última instancia, la validación de este modelo de ratón procederá de experimentos en los que se mida la fuerza y la velocidad de deslizamiento de la ß-MyHC humana y de los filamentos finos reguladores del corazón. Las investigaciones que se realicen usando estos modelos deberían ayudar a mejorar el manejo clínico de la MHF en humanos, y a establecer dianas terapéuticas y/o intervenciones diseñadas para enlentecer o revertir el desarrollo del fenotipo hipertrófico de la MHF y otras miocardiopatías (fig. 1).

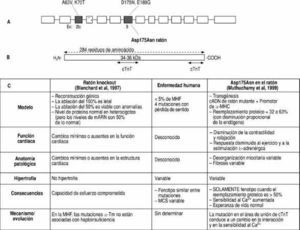
Fig. 1. Las mutaciones de la cadena pesada b de la miosina en la MHF. (A) Se indican las mutaciones humanas previamente descritas, el color rojo se refiere a las más malignas. La organización intrónexón del gen está representada y los exones afectados aparecen en azul. Las mutaciones que han sido obtenidas en modelos animales genéticamente modificados se muestran debajo del diagrama del intrónexón. (B) Representación esquemática de la proteína b-MHC codificada por el gen MYH7,con sus dominios y sus lugares de unión. (C) Características principales de los modelos animales que reproducen algunas de las mutaciones humanas, comparación con la enfermedad humana y la patogenia de la enfermedad. Las mutaciones con pérdida de sentido están representadas por la letra que codifica el residuo de aminoácido (aa) que está mutado seguido por el número de la posición del aa, seguido por la letra del nuevo aa. Las mutaciones por truncamiento pueden ser consecuencia de mutaciones del lugar de cor-te y empalme (Splx ; donde x es la posición de la mutación del nucleótido), mutaciones por deleción (dely o D1-y; donde y es la posición numérica del último residuo de aminoácido presente en la proteína truncada), y mutaciones con desplazamiento de marco (Framz ; donde z es la posición de la mutación del nucleótido). * indica una mutación de sustitución compleja que afecta a los genes MYH6y MYH7. Ex: exón; In: intrón.
¿Hasta qué punto es válido extrapolar los resultados obtenidos en ratones a los humanos? Para abordar esta cuestión, Roopnarine et al63 colocaron 3 mutaciones humanas en la *-MyHC de roedor y examinaron las actividades de la ATPasa. Curiosamente, el grado de afección de la enzima de las miosinas mutantes se correlacionó con el fenotipo clínico de los pacientes que tenían la correspondiente mutación. Estos datos sugieren que, a pesar de las diferencias entre especies, los modelos animales pueden ser perfectamente predictivos para los humanos. Otro enfoque para responder esta pregunta es generar animales más grandes que contengan el locus de la enfermedad. Hasta el momento, la mutación Arg403Gln se ha generado también en el conejo que, como en humanos, y a diferencia del ratón, expresa la misma isoforma ß-MyHC cardíaca (las 2 proteínas comparten un 98% de homología). Los conejos transgénicos que sobreexpresaron la forma humana mutante de la ß-MyHC en el corazón, y que tuvieron una sustitución parcial de la proteína normal con el polipéptido mutante, presentaron hipertrofia cardíaca, desorganización miocítica y miofibrilar y un aumento en el contenido de colágeno intersticial, pero tuvieron una función sistólica normal3. Se produjo una gran incidencia de muerte prematura, y el fenotipo observado fue muy similar al de los humanos. La proteína mutante ejerció un efecto negativo dominante, lo que concuerda con la genética de la población humana. Por lo tanto, el conejo transgénico que expresa la proteína mutante Arg403Gln es un modelo atractivo para el estudio de la patogenia y la búsqueda de dianas terapéuticas de la miocardiopatía hipertrófica humana. El conejo es también un modelo más apropiado para realizar estudios funcionales no invasivos con ecocardiografía o electrofisiología. Aunque estas técnicas han sido desarrolladas para utilizarse en el ratón64, siguen estando restringidas a un pequeño número de centros académicos especializados, y el modelo de conejo llegará a ser, con toda seguridad, más accesible a la comunidad cardiológica general para un estudio de tallado. En concreto, ya se han realizado estudios electrofisiológicos extensivos en el conejo65, y han aportado datos útiles sobre la disfunción arritmogénica que es común en la MHF, especialmente la ocurrencia de muerte cardíaca súbita.
Las cadenas ligeras esencial (ELC) y reguladora de la miosina (RLC)
Dos únicos tipos de cadenas ligeras de la miosina se unen a la región del cuello (región reguladora) de la cabeza de la miosina: la esencial (MLC1 o ELC) y la reguladora (MLC2 o RLC). Estas pequeñas proteínas parecen desempeñar papeles estructurales y reguladores en la función de la miosina, estabilizando el largo cuello *-helicoidal de la cabeza de la miosina y afectando su rigidez o dureza66. Pertenecen a la superfamilia de las proteínas EF-hand, que incluye la calmodulina y la troponina C. Las isoformas específicas de mi compartimiento cardíaco se expresan en la mayoría de vertebrados, incluida la especie humana. La cadena ligera esencial (o álcalis) de la miosina ventricular contiene 195 residuos de aminoácidos (PM aproximado de 21-25 KDa), está codificada por el gen MYL3 localizado en el cromosoma 3p21.2-p21.3, constituido por 7 exones (fig. 2). La cadena ligera reguladora está compuesta por 166 residuos de aminoácidos (PM aproximado de 25-37 Kda), está codificada por el gen MYL2 localizado en el cromosoma 12q23-q24.3, con 7 exones (fig. 2). La MLC2 puede ser fosforilada a través de la vía de la proteína quinasa A.

Fig. 2. Las mutaciones de la cadena ligera de la miosina en la MHF. (A) Se indican las mutaciones humanas previamente descritas. La organización intrón-exón del gen se representa y los exones afectados se muestran en azul. Las mutaciones que han sido obtenidas en modelos animales genéticamente modificados se muestran debajo del diagrama del intrón-exón. (B) Representación esquemática de las proteínas ELC y RLC codificadas por los respectivos genes MYL3 y MYL2, con los lugares de unión importantes. (C) Características principales de los modelos animales que reproducen algunas de las mutaciones humanas, comparación con la enfermedad humana y la patogenia de la enfermedad. Las mutaciones con pérdida de sentido están representadas por la letra que codifica el residuo de aminoácido (aa) que está mutado seguido por el número de la posición del aa, seguido por la letra del nuevo aa. Las mutaciones por truncamiento como las mutaciones del lugar de corte y empalme aparecen como Splx; donde x es la posición de la mutación del nucleótido. Ex: exón; In: intrón.
El concepto de que las mutaciones en las cadenas ligeras de los miosina pueden causar la MHF apareció como consecuencia de los numerosos resultados que apuntaban a que las alteraciones en otras proteínas sarcoméricas podían conducir a la enfermedad. Además, un grupo importante de mutaciones de la MyHC se había mapeado en los dominios implicados en la unión a las cadenas ligeras, lo que implicaba a esta región como una zona crítica para el funcionamiento normal de las proteínas motoras. Actualmente se han identificado mutaciones puntuales tanto en la ELC como en la RLC. Existen 2 mutaciones con pérdida de sentido en la ELC en los residuos conservados de aminoácidos Met149Val y Arg154His que están asociadas a la enfermedad67,68. Los estudios de estructura/función demuestran que existe un aumento en la velocidad de translocación de la actina en ensayos de motilidad in vitro67,68. También se han identificado 7 mutaciones con pérdida de sentido y una mutación por truncamiento en la RLC67,69. Más de la mitad de estas mutaciones (Ala13Thr, Phe18Leu, Glu22Lys, Pro94Arg) tuvieron lugar en residuos de aminoácidos muy conservados, y mediante modelos animales se demostró que estas mutaciones pueden producir la eliminación del lugar de fosforilación. Esto, a su vez, puede tener efectos sobre la flexibilidad del cuello de la miosina. Tal como ocurre con las mutaciones de la MyHC, las mutaciones de la RLC muestran una expresión variable y una penetrancia incompleta.
Las 2 mutaciones de la cadena ligera están asociadas con un fenotipo cardíaco raro y sorprendente, que se acompaña por una hipertrofia masiva de los músculos papilares y del tejido ventricular adyacente, causando una obstrucción medioventricular67. Las mutaciones de la MLC pueden interferir con la respuesta de estiramiento-activación del músculo papilar y del tejido ventricular adyacente, una propiedad que sólo se ha encontrado en porciones del corazón que aumentan su potencia70. Estos cambios se pueden traducir en un empeoramiento de la elasticidad de la región del cuello de la miosina, dando lugar a una reducción en la capacidad oscilatoria de los músculos papilares, que normalmente está aumentada por una fuerte respuesta de estiramiento-activación. Los autores han hipotetizado que, con el tiempo, se produce una respuesta hipertrófica compensatoria para incrementar la potencia que finalmente obstruye la cavidad ventricular.
Con la intención de validar la hipótesis de que la hipertrofia del músculo papilar, asociada con las mutaciones de cualquiera de las cadenas ligeras, refleja un efecto común sobre la función de la miosina, se han generado unos ratones transgénicos con unos fragmentos genómicos humanos grandes (de aproximadamente 12 kilobases) que contienen el locus mutado para la Met149Val de la cadena ligera de la miosina. El fenotipo fue reproducido70. Este modelo ha proporcionado una oportunidad para estudiar la respuesta de estiramiento-activación antes de que los corazones fueran deformados por el proceso hipertrófico, y ha confirmado que la respuesta de estiramiento-activación puede desempeñar un papel en el corazón de los mamíferos, ofreciendo una nueva vía para modular la función cardíaca humana. Además, un modelo novedoso de ratón transgénico en el que el lugar susceptible de ser fosforilado es eliminado ha demostrado la importancia de esta modificación que tiene lugar después de la traducción71. De hecho, la modificación farmacológica de la fosforilación de la RLC cardíaca puede representar una nueva diana terapéutica en la insuficiencia car díaca.
Como se ha mencionado anteriormente, los ratones transgénicos ELC se crearon con un fragmento genómico humano intacto. Así, aunque el fenotipo fue fielmente reproducido, el experimento estuvo sujeto a cierta ambigüedad en relación con el papel causativo de la mutación Met149Val, ya que muchas otras secuencias estuvieron presentes. Para establecer sin ambigüedades la relación causal para las mutaciones de las cadenas ligeras reguladora y esencial en la hipertrofia cardíaca, se crearon múltiples líneas de ratones que expresaban o bien las formas mutadas o bien las formas originales, utilizando clones de cADN que contenían sólo las regiones codificadoras de los loci genéticos. Sorprendentemente, cuando niveles importantes de expresión resultaban en un reemplazamiento de la proteína endógena por la forma mutada igual o mayor a un 50%, no se detectó hipertrofia, incluso en animales de edad avanzada72. Aunque los cambios ocurrieron a nivel de los miofilamentos y de las células, y las miofibrillas mostraban una sensibilidad aumentada al calcio y déficit significativos en la relajación de forma relacionada con la dosis transgénica, no apareció hipertrofia franca ni en los cardiomiocitos ni en las cámaras. Debido a la discordancia entre estos datos y los obtenidos en ratones transgénicos que contenían el fragmento genómico humano, la opinión actual de que estas mutaciones puntuales causan por sí mismas hipertrofia cardíaca significativa debe ser reevaluada. Queda la posibilidad de que estas mutaciones puntuales puedan estar simplemente ligadas de forma estrecha a la lesión real, aunque todavía por descubrir, en el locus humano.
Estos resultados demuestran la utilidad de la transgénesis y de los modelos animales para establecer las relaciones únicas entre estructura y función en las proteínas mutadas. El remodelado genético permite a la comunidad científica probar las relaciones causales de mutaciones particulares. Este tipo de experimentos tiene implicaciones significativas para la práctica clínica, ya que antes de que se establezcan mejor los procesos de muestreo, es imperativo conocer la relación directa entre causa y efecto para una mutación particular.
La proteína C que se une a la miosina (MyBP-C)
La MyBP-C es una gran proteína (PM aproximado de 130KDa) de 1.173 residuos de aminoácidos. Teniendo en cuenta su abundancia en el cardiomiocito (puede llegar a constituir el 4% de la masa proteica del cardiomiocito) y que han transcurrido 25 años desde que se descubrió, su función (o funciones) han permanecido sorprendentemente poco claras. La MyBP-C, el principal componente del filamento grueso, se une tanto a los sistemas de filamentos de la miosina (filamentos gruesos) como a los de titina. Después de su descubrimiento hace 27 años73, el interés en el papel de esta proteína se ha intensificado al haberse establecido una relación entre la aparición de mutaciones múltiples en su estructura polipeptídica y la MHF31. La MyBP-C se encuentra localizada en la región C de la banda A, y tiene una organización única, con 7-9 bandas axiales en cada mitad sarcomérica. Como otras proteínas que se ligan a la miosina, la MyBP-C pertenece a la superfamilia de inmunoglobulinas (Ig) intracelulares, y está compuesta por dominios Ig y fibronectina repetidos74. Los modelos in vitro y los experimentos de reconstitución, así como los experimentos llevados a cabo usando transfecciones celulares, apuntan a que la proteína probablemente desempeña un papel importante en el ensamblaje y el mantenimiento general de la arquitectura del sarcómero75,76. La isoforma cardíaca, que consiste en 35 exones (fig. 3), está codificada por el gen humano MYBPC3, localizado en el cromosoma 11p11.2. La MyBP-C cardíaca contiene 3 dominios únicos de isoforma, algunos de los cuales pueden modular la contracción vía fosforilación por proteína quinasa A y/o proteína quinasa dependiente de calmodulina.

Fig. 3. Las mutaciones de la proteína C que se une a la miosina (MyBP-C) en la MHF. (A) Se indican las mutaciones humanas previamente descritas. La organización intrón-exón del gen se representa y los exones afectados se muestran en azul. Las mutaciones que han sido obtenidas en modelos animales genéticamente modificados se muestran debajo del diagrama del intrón-exón. (B) Representación esquemática de la proteína MyBP-C codificada por el gen MYBPC3, con sus dominios y lugares de unión. (C) Características principales de los modelos animales que reproducen algunas de las mutaciones humanas, comparación con la enfermedad humana y la patogenia de la enfermedad. Las mutaciones con pérdida de sentido están representadas por la letra que codifica el residuo de aminoácido (aa) que está mutado seguido por el número de la posición del aa, seguido por la letra del nuevo aa. Las mutaciones por truncamiento pueden derivarse de mutaciones del lugar de corte y empalme (Splx; donde x es la posición de la mutación del nucleótido), mutaciones por deleción (dely o 1-y; donde y es la posición numérica del último residuo de aminoácido presente en la proteína truncada), y mutaciones con desplazamiento de marco (Framz; donde z es la posición de la mutación del nucleótido).
Las mutaciones en el gen de la MyBP-C cardíaca son responsables de aproximadamente el 15% de los casos de MHF. Se han descrito un gran número de mutaciones (> de 31), la mayoría de las cuales resultan en mutaciones del lugar de corte y empalme, inserciones y deleciones31,77-80, que producen proteínas truncadas. La mayoría de los truncamientos ocurren en el dominio terminal COOH, que contiene un lugar de unión para la miosina, estando presente unas veces sí y otras no el lugar de unión para la titina. Curiosamente, la presentación de la enfermedad no se produce hasta la quinta-sexta década de vida, y se caracteriza por un comienzo tardío de la MHF, penetrancia incompleta y un perfil clínico relativamente benigno81, que contrasta con otras mutaciones genéticas sarcoméricas más malignas. Debido a estas características, los cambios patológicos longitudinales ligados a la enfermedad en los pacientes con alteraciones en la MyBP-C son difíciles de obtener.
El enfoque transgénico debería proporcionar un instrumento poderoso para estudiar los procesos patológicos longitudinales relacionados con el comienzo de la enfermedad y su progresión en edades más avanzadas. Esta aproximación experimental puede también ayudar a determinar las relaciones básicas entre estructura y función, y establecer la curva dosis-respuesta por la que el comienzo, la severidad y la progresión de la enfermedad puede correlacionarse con los niveles de la proteína mutada. De hecho, la transgénesis proporciona al investigador la tecnología para distinguir entre 2 causas diferentes fundamentales de la enfermedad: el efecto debido al «péptido tóxico» o la haploinsuficiencia funcional79. En esta enfermedad, la haploinsuficiencia fue implicada por el hallazgo de que la proteína mutada no estaba presente en las biopsias humanas80. Esto sugirió la posibilidad de que la proteína truncada era inestable y la enfermedad se desarrollaba no por la presencia del polipéptido mutado sino porque había una cantidad insuficiente de la proteína normal.
Para probar esta posibilidad, así como para crear modelos en los que los procesos patológicos puedan ser discernidos a lo largo de la vida del animal, se han generado 2 modelos de ratones transgénicos. El primer modelo reemplazó aproximadamente la mitad de la MyBP-C cardíaca con una proteína que carecía de los dominios para unirse a la miosina y la titina82. La proteína truncada que se codificaba era estable, pero no se podía incorporar de manera eficiente en el sarcómero, lo que sugería que la proteína mutante no actúa como un «péptido tóxico». Las anormalidades fisiopatológicas y ultraestructurales en los ratones estaban relacionadas con la dosis, e ilustraban las consecuencias estructurales causadas por la insuficiente incorporación de MyBP-C funcional en el ensamblaje sarcomérico y/o por la incorporación de polipéptido aberrante. La disgenesia sarcomérica era prevalente, lo que indica que cantidades normales de la proteína original son necesarias para la integridad estructural de los sarcómeros de los cardiomiocitos. El fenotipo de estos ratones fue sorprendentemente leve durante el desarrollo y el inicio de la edad adulta, reproduciendo el fenotipo clínico relativamente benigno. La función orgánica se mantuvo inalterada en los ratones adultos jóvenes, mientras que la mecánica de las fibras era compatible con alteraciones sutiles en la generación de fuerza. Además, de acuerdo con los datos clínicos, el proceso hipertrófico era muy leve.
Sin embargo, los mecanismos fisiopatológicos que son relevantes para este fenotipo pueden variar considerablemente, dependiendo de la mutación particular de la MyBP-C. Para probar esta hipótesis, se ha generado un segundo modelo de ratón transgénico que expresa en el corazón una forma mutante de la MyBP-C que carece únicamente del lugar de unión para la miosina83. Tal como se esperaba, esta mutación exhibió unas características muy diferentes. Sólo niveles modestos de proteína se encontraron en el corazón, lo que es consistente con los datos procedentes de biopsias humanas80. A pesar de haber niveles normales de la MyBP-C endógena, la disgenesia sarcomérica estuvo también presente a nivel ultraestructural en este modelo, lo que confirma que para esta mutación particular, la proteína mutada actúa más bien como un «péptido tóxico». De acuerdo con las observaciones clínicas, el fenotipo del órgano entero y del animal fue bastante sutil y difícil de detectar antes de un año, aunque hubo una ligera hipertrofia, y la mecánica de las fibras estaba dañada en estadios tempranos. Sin embargo, los
ratones transgénicos presentaron una tolerancia comprometida al ejercicio asociada con bradicardia, reminiscente de la respuesta hipotensiva que se había encontrado en pacientes con MHF que habían practicado ejercicio84. Estos 2 modelos se encuentran comparados y contrastados en la figura 3.
Los modelos transgénicos de MyBP-C ilustran algunos de los puntos importantes concernientes a la utilidad del enfoque general. En primer lugar, un modelo animal de roedor pequeño puede reproducir fielmente elementos de la patología humana. En segundo lugar, la tasa relativamente rápida con la que estos modelos pueden ser generados permite mostrar la diversidad de los mecanismos fisiopatológicos que pueden resultar en una MHF, en la que están involucradas diferentes mutaciones del mismo gen (MYBPC3). Debido al número limitado de investigaciones que pueden realizarse en humanos, los modelos de ratón proporcionan una oportunidad única para descubrir procesos patogénicos que evolucionan gradualmente a lo largo de la vida del animal.
Alfa-tropomiosina (*-Tm)
La tropomiosina (Tm) es una proteína rígida en forma de bastón que se une a todo lo largo del filamento de actina y al complejo de troponina. Regula la in teracción sensible al calcio de la actina y la miosina. En el corazón adulto, la isoforma predominante es la *-Tm de músculo estriado, compuesta por 284 residuos de aminoácidos (PM aproximado de 34-36 KDa). Un mecanismo alternativo de corte y empalme del gen TPM1 genera *-Tm. El gen humano está localizado en el cromosoma 15q22 y consiste en 14 exones (fig. 4). La *-Tm contiene 2 dominios de unión TnT, uno dependiente de calcio y otro independiente de calcio en la región COOH-terminal de la molécula que une la *-Tm al complejo de troponina. La *-Tm estabiliza el filamento y lo hace más rígido y, en ausencia de calcio, bloquea el lugar de unión de la actina a la miosina. También desempeña un papel a la hora de determinar el grado de cooperatividad y sensibilidad al calcio.

Fig. 4. Las mutaciones de la *-tropomiosina (*-Tm) en la MHF. (A) Se indican las mutaciones humanas previamente descritas. La organización intrón-exón del gen se representa y los exones afectados se muestran en azul. Las mutaciones que han sido obtenidas en modelos animales genéticamente modificados se muestran debajo del diagrama del intrón-exón. (B) Representación esquemática de la proteína *-Tm codificada por el gen TPM1, con sus lugares de unión. (C) Características principales de los modelos animales que reproducen algunas de las mutaciones humanas, comparación con la enfermedad humana y la patogenia de la enfermedad. Las mutaciones con pérdida de sentido están representadas por la letra que codifica el residuo de aminoácido (aa) que está mutado seguido por el número de la posición del aa, seguido por la letra del nuevo aa. Ex: exón.
Las mutaciones en el gen TPM1 representan < 5% de las mutaciones responsables de MHF. Se han identificado 4 mutaciones con sentido erróneo, 2 en el exón 2 (Ala63Val, Lys70Thr) que pueden alterar la unión de la *-Tm a la actina85,86, y 2 en el exón 5 (Asp175Asn, Glu180Gly) que pueden afectar la unión a la TnT dependiente de calcio87,88. La presentación clínica asociada a las diferentes mutaciones es bastante similar85, pero la severidad fenotípica no está bien establecida. Dependiendo de la población étnica, la importancia de la hipertrofia del ventrículo izquierdo que puede conducir a una dilatación progresiva del ventrículo izquierdo y muerte cardíaca súbita, varía sustancialmente. Hasta muy recientemente, la mutación Asp175Asn era la más estudiada: se han usado pruebas de expresión de proteína mutante e incorporación en el sarcómero utilizando músculos de pacientes, y se ha confirmado que la mutación actúa como un alelo negativo dominante más que como un «alelo nulo»88.
Para estudiar las consecuencias funcionales de las mutaciones de Tm, se han generado ratones transgénicos que expresan la mutación con sentido erróneo Asp175Asn en el compartimiento cardíaco89. La expresión de la proteína mutante estuvo asociada con una disminución recíproca en los niveles endógenos de *-Tm. Cuando el reemplazo excedía el 50% de la *-Tm endógena, se producía una disminución de la contractilidad y relajación en preparaciones de corazón funcionante. Mediante ecocardiografía se pudo ver que los ratones transgénicos exhibían una función ventricular normal, pero respondían menos vigorosamente al ejercicio y a la estimulación adrenérgica de tipo ß. Sin embargo, la esperanza de vida era normal, tal como ocurre en algunos pacientes. Histológicamente se apreciaba una ocurrencia variable de desorganización miocítica, hipertrofia y fibrosis. La mutación, que está localizada en el área de unión de la *-Tm a la TnT, puede deteriorar las ya de por sí débiles interacciones TnT-*-Tm que se producen en esta región, alterando la sensibilidad de los miofilamentos al calcio. Para probar esta hipótesis, se aislaron fibras de músculos papilares a las que se eliminó la membrana celular. La mecánica y cinética de estas fibras, tanto de los corazones de ratón mutante como de los músculos humanos, mostraron una sensibilidad aumentada al calcio88.
Los datos complementarios esenciales para ayudar a entender la patogenia de la MHF relacionada con las mutaciones de la *-Tm proceden de un modelo de ratón «knockout» en el que el gen de la *-Tm fue eliminado por reconstrucción génica90. Tal como se esperaba, los ratones «knockouts» homocigotos no fueron viables. Sin embargo, los ratones heterocigotos apenas presentaron cambios en la estructura o función cardíacas, lo que demuestra que el ratón puede afrontar fácilmente la haploinsuficiencia del gen de la *-Tm. Estos datos sugieren fuertemente que las anormalidades cardíacas debidas a mutaciones de la *-Tm no están asociadas a haploinsuficiencia, como ocurre con las ablaciones del gen de la *-MyHC18, siempre que asumamos que los músculos cardíacos humano y de ratón son similares. Es interesante señalar que el ARNm total estaba disminuido en un 50% en los ratones heterocigotos. Sin embargo, el ARNm unido a los polisomas (es decir, el que se traduce activamente a proteína), así como los niveles de proteína, fueron similares a los que presentaron las camadas del tipo original, lo que sugiere que la cantidad relativa de ARNm que se traduce no está afectada, y que la regulación de la traducción desempeña un papel importante en el control de la expresión de la Tm91. Estos hallazgos implican que en los heterocigotos los mecanismos reguladores mantienen el nivel de Tm miofibrilar a pesar de la reducción en el ARNm de la *-Tm, y explican por qué la inactivación de uno de los alelos de la *-Tm no causa la patología observada en la MHF (fig. 4).
Troponina T cardíaca
La TnT cardíaca (cTNT: PM aproximado de 37KDa), la subunidad que se une a la tropomiosina del complejo de troponina, es una molécula asimétrica de 294 residuos de aminoácidos. En el corazón humano se han caracterizado 4 isoformas: cTnT1-cTnT492,93; su expresión depende del estadio de desarrollo y de las condiciones fisiopatológicas. Todas las isoformas cardíacas humanas derivan de un mecanismo alternativo de corte y empalme de un único gen, el TNNT2, localizado en el cromosoma 1q3287 y compuesto por 17 exones (fig. 5). La TnT cardíaca tiene varios dominios: la región NH2-terminal contiene un lugar de fosforilación que es diana de la proteína quinasa C, así como un lugar de unión para la región C-terminal de la *-Tm. La región COOH terminal contiene un lugar de unión para la *-Tm dependiente de calcio y lugares
de unión para la TnC, TnI y, posiblemente, para la actina. Existen 3 lugares potenciales de fosforilación, que pueden regular la cinética de entrecruzamiento al disminuir la tasa máxima de actividad de la ATPasa94. La proteína desempeña un papel estructural importante al colocar el complejo de troponina a lo largo del filamento fino. Además, la TnT confiere sensibilidad al calcio a la actividad inhibitoria del complejo cTnI-cTnC sobre la ATPasa de la actomiosina95.

Fig. 5. Las mutaciones de la troponina T cardíaca (cTnT) en la MHF. (A) Se indican las mutaciones humanas previamente descritas. La organización intrón-exón del gen se representa y los exones afectados se muestran en azul. Las mutaciones que han sido obtenidas en modelos animales genéticamente modificados se muestran debajo del diagrama del intrón-exón. (B) Representación esquemática de la proteína cTnT codificada por el gen TNNT2, con sus lugares de unión. (C) Características principales de los modelos animales que reproducen algunas de las mutaciones humanas, comparación con la enfermedad humana y la patogenia de la enfermedad. Las mutaciones con sentido erróneo están representadas por la letra que codifica el residuo de aminoácido (aa) que está mutado seguido por el número de la posición del aa, seguido por la letra del nuevo aa. Las mutaciones por truncamiento pueden derivarse de mutaciones del lugar de corte y empalme (Splx; donde x es la posición de la mutación del nucleótido), mutaciones por deleción (dely o 1-y; donde y es la posición numérica del último residuo de aminoácido presente en la proteína truncada). Ex: exón; In: intrón.
Las mutaciones en el gen TNNT2 son responsables del 15% de las MHF96-98. Hasta la fecha se han descrito 14 mutaciones; 12 son mutaciones con pérdida de sentido distribuidas a lo largo de la molécula, 1 es una mutación del lugar de corte y empalme87 y 1 es una deleción en marco96,99. Estas 2 últimas producen una proteína truncada. A pesar de esta diversidad, la presentación clínica es normalmente muy similar, caracterizada por una hipertrofia indetectable, suave o moderada, una penetrancia incompleta, un pronóstico pobre y una alta incidencia de muerte cardíaca súbita en la adolescencia o en el inicio de la edad adulta. En esta clase altamente maligna el desarrollo de hipertrofia cardíaca puede ocurrir independientemente de la muerte súbita, lo que distingue este fenotipo de las mutaciones de la proteína motora, la miosina.
Se han simulado experimentalmente 2 mutaciones humanas diferentes de la cTnT: una mutación del lugar de corte y empalme encontrada en el intrón 15 del ADN genómico87, y una mutación con pérdida de sentido Arg92Gln87. Los ratones transgénicos que expresaban la mutación del lugar de corte y empalme produjeron, según lo previsto, una proteína cTnT truncada en el dominio COOH-terminal100. Desarrollaron miocardiopatía y exhibieron desorganización miofibrilar y disfunción diastólica y sistólica leve. Los corazones mutantes fueron más pequeños, debido a una menor hiperplasia y a un menor tamaño de los cardiomiocitos. A partir de este modelo se obtuvo una mejor comprensión de la patogenia de esta enfermedad, ya que se comprobó que la proteína truncada era incorporada en el sarcómero. Tal como ocurría con la MyBP-C inestable, la cTnT mutada estaba presente sólo a muy bajos niveles (< 5% de la cTnT total), lo que sugiere un mecanismo de «péptido tóxico» más que un «alelo nulo». Además, la presencia en el corazón de niveles más elevados de la proteína mutante (> 10%) producía la muerte en las primeras 24 horas de vida, lo que subraya la relación dosis-efecto entre la cantidad de cTnT mutante y la mortalidad temprana. Así pues, la severidad del fenotipo en los animales debería considerarse en comparación con la alta incidencia de muerte cardíaca súbita observada en pacientes, lo que puede estar relacionado con el nivel de proteína truncada expresada en la enfermedad.
Para intentar resolver la diversidad de mecanismos que conducen a la enfermedad humana, se han reproducido en 3 modelos de ratones los efectos de una mutación diferente de la cTnT que corresponde a la mutación con pérdida de sentido Arg92Gln. Estos modelos, que expresaron niveles variables de la proteína mutada, exhibieron fenotipos diferentes, pero presentaron una característica común: la ausencia de hipertrofia. En consecuencia, la cantidad de proteína mutada puede desempeñar un papel importante a la hora de determinar el fenotipo de la enfermedad, y puede estar involucrada en la presentación heterogénea observada en una población de pacientes que tienen una mutación idéntica (alternativamente, otros genes modificadores pueden ser responsables de la presentación variable). Cuando sólo se expresaban niveles bajos de la proteína mutante, el corazón del ratón presentaba una desorganización de los miocitos cardíacos, un aumento del contenido intersticial de colágeno y una disfunción diastólica101, fenotipos similares a los encontrados en la MHF humana. En estos casos, la enfermedad humana ocurre probablemente a través de un efecto nega tivo dominante en el cual la función alterada de los cardiomiocitos y la desorganización son las anormalidades primarias, y la hipertrofia un proceso compensatorio. Cuando se obtuvieron niveles más altos de expresión de la proteína mutante en dos laboratorios diferentes, los ratones transgénicos presentaron un fenotipo distinto, en el cual el deterioro de la función cardíaca global era un efecto primario y tenía lugar independientemente del desarrollo de las anormalidades histológicas de la MHF. Aparte de este mecanismo de acción crucial y común, cada uno de los 2 modelos transgénicos presentó unas características distintivas propias, que pueden reflejar la expresión clínica heterogénea de la enfermedad, debida a diferencias en el fundamento y/o factores del entorno. Los ratones transgénicos que sobreexpresan el cADN mutante humano controlado por el potente promotor de *-MyHC de ratón102 mostraron una disminución de la fracción de eyección del ventrículo izquierdo (LVEF), en contraste con la LVEF normal que se había encontrado en pacientes humanos. Este tipo de discordancia puede representar también uno de los límites en el uso de modelos animales genéticamente modificados, y en la extrapolación de los resultados desde el ratón a la enfermedad humana. En otro modelo de ratón transgénico, que utilizó un cADN mutante de ratón controlado por el promotor de la *-MyHC de rata103, niveles elevados de la proteína mutante (30, 67 y 92% del total de cTnT) estuvieron asociados con alteraciones dependientes de la dosis. Estas alteraciones incluyeron ventrículos izquierdos más pequeños, una inducción significativa del factor natriurético atrial y transcriptos ß-MyHC en los corazones (un patrón transcripcional característico de la respuesta hipertrófica), y patología mitocondrial. Sin embargo, cuando son expresados niveles elevados de la cTnT mutante, la mutación puede actuar como un «cambio en la función» más que como un «péptido tóxico», lo que explica las diferencias fenotípicas observadas entre modelos que exhiben varios niveles de la proteína mutante. En cada uno de estos modelos, es importante enfatizar que la estoiquiometría global de la proteína no estaba modificada. Es decir, la cantidad de cTnT mutante + cTnT endógena equivalía a los niveles de cTnT observados en los animales normales no modificados, lo que indica que la forma endógena es reemplazada por la cTnT mutante en grado variable.
En conjunto, estos modelos de ratón demuestran que los diferentes alelos de la cTnT están asociados con fenotipos distintos, lo que sugiere que la heterogeneidad clínica de la MHF es, al menos parcialmente, debida a mecanismos específicos de los alelos (fig. 5). En el futuro, la información procedente de los modelos de ratón modificados por ingeniería genética será complementada por estudios de terapia génica. Esto permitirá la expresión temporal de una proteína mutada en el corazón. Recientemente, Yu et al han expresado la cTnT mutante Arg92Gln en el corazón adulto de conejo mediante una inyección intramiocárdica directa de los adenovirus recombinantes104. En contradicción con los estudios a largo plazo in vivo y a corto plazo in vitro, la proteína mutante se incorporó en el sarcómero sin inducir una desorganización temprana en los miocitos y en los sarcómeros, lo que sugiere que la incorporación miofibrilar prolongada de la cTnT mutante es esencial para su efecto negativo dominante sobre la estructura de los miocitos cardíacos en el miocardio intacto.
Troponina I cardíaca
La TnI es la subunidad inhibitoria del complejo de troponina. El corazón tiene una isoforma cardíaca específica que se caracteriza por tener una extensión NH2-terminal de 33 residuos de aminoácidos que no tienen las isoformas esqueléticas. La TnI se une a la actina, lo que previene la actividad ATPasa de la miosina. La inhibición por esta subunidad se vuelve reversible en presencia de calcio, que se une a la cTnC e induce un cambio conformacional en el complejo de la troponina que produce la liberación de la actividad inhibitoria de la cTnC, permitiendo la contracción. La isoforma cardíaca presenta unas propiedades únicas, que incluyen una unión cooperativa a la actina-tropomiosina105. El polipéptido tiene 210 aminoácidos (PM aproximado de 30KDa) y es codificado por el gen TNN13, localizado en el cromosoma 19p13.2-q13.2, y compuesto por 8 exones (fig. 6). La TnI cardíaca contiene varios dominios funcionales. En la extensión que se encuentra en el extremo NH2-terminal hay 2 lugares que pueden ser fosforilados por la proteína quinasa A (serinas en posición 23 y 24). La fosforilación disminuye la sensibilidad de los miofilamentos al calcio, e inhibe la unión cooperativa a la actina. Las serinas que están en las posiciones 42 y 44 pueden ser fosforiladas por la proteína quinasa C: la fosforilación reduce la actividad máxima ATPasa. Una región inhibitoria que se une a la actina y a la cTnC produce relajación a través de la inhibición de la interacción con la actomiosina. Por último, una región COOH-terminal parece ser esencial para la sensibilidad de los miofilamentos al calcio106.

Fig. 6. Las mutaciones de la troponina I cardíaca (cTnI) en la MHF. (A) Se indican las mutaciones humanas previamente descritas. La organización intrón-exón del gen se representa y los exones afectados se muestran en azul. Las mutaciones que han sido obtenidas en modelos animales genéticamente modificados se muestran debajo del diagrama del intrón-exón. (B) Representación esquemática de la proteína cTnI codificada por el gen TNNI3, con sus lugares de unión. (C) Características principales de los modelos animales que reproducen algunas de las mutaciones humanas, comparación con la enfermedad humana y la patogenia de la enfermedad. Las mutaciones con sentido erróneo están representadas por la letra que codifica el residuo de aminoácido (aa) que está mutado seguido por el número de la posición del aa, seguido por la letra del nuevo aa. Ex: exón.
Las mutaciones de la cTnI también están asociadas con la MHF107. Hasta la fecha se han descrito 8 mutaciones, 7 mutaciones con pérdida de sentido y una deleción, todas localizadas entre los exones 5 y 8. Tres mutaciones (Arg145Gly, Arg145Gln y Arg162Trp) se encuentran en la región inhibitoria, y las otras están en la región COOH de la molécula. Las consecuencias funcionales de todas estas mutaciones siguen siendo desconocidas. Producen una hipertrofia ventricular que, en algunos casos, puede estar restringida al ápex107.
En un intento por elucidar los mecanismos por los que las mutaciones en la cTnI pueden producir el desarrollo de MHF, se han realizado análisis in vitro utilizando complejos de troponina cardíaca humana reconstituida que expresan las formas mutantes de cTnI: Arg145Gly y Arg136Trp. Estas 2 mutaciones disminuyeron la inhibición de la actividad ATPasa bajo condiciones de relajación, y aumentaron la sensibilidad de la enzima al calcio. In vivo esto se puede traducir en un empeoramiento de la relajación y una contractilidad alterada, lo que puede constituir el estímulo hipertrófico que conduce a una MHF108. James et al109 también han probado el mecanismo patogénico de la enfermedad in vivo generando ratones transgénicos que contienen la mutación con pérdida de sentido Arg145Gly. Los animales desarrollaron una patología similar a la MHF humana, con hipertrofia en los cardiomiocitos y fibrosis intersticial. Las alteraciones funcionales a nivel del órgano entero incluyeron hipercontractilidad con disfunción diastólica, y se caracterizaron a nivel de la fibra por un aumento en la sensibilidad al calcio. Sin embargo, el fenotipo en los animales fue mucho más severo, con aparición frecuente de muerte cardíaca súbita y un acortamiento drástico en la esperanza de vida. Esta diferencia puede ser atribuida a las características del corazón de ratón, que tiene que ciclar muy rápidamente de sístole a diástole, y tiene que ser entonces más sensible (que el corazón humano) a las perturbaciones en el manejo del calcio (fig. 6).
La sensibilidad del corazón de ratón a las proteínas anormales de troponina está apoyada por las observaciones obtenidas a partir de mutaciones en múltiples subunidades de troponina cardíaca (cTnT, cTnI). Los modelos de ratón proporcionan una herramienta importante para entender las relaciones patogénicas resultantes entre estructura y función y subrayar las diferencias en la severidad del fenotipo de las mutaciones de troponina entre el corazón humano y el de ratón.
Miocardiopatía dilatada familiar
La miocardiopatía dilatada familiar (MDF) es una enfermedad genética autosómica dominante que representa el 20-30% de las miocardiopatías dilatadas idiopáticas (MCD), que son responsables del 60% de todas las miocardiopatías. Las miocardiopatías dilatadas idiopáticas son una causa importante de morbilidad y mortalidad, y una causa común de insuficiencia car díaca que conduce al trasplante cardíaco. La MDF se caracteriza por una dilatación ventricular y una disminución de la función sistólica; el diagnóstico normalmente se basa en la ecocardiografía. El desarrollo de muerte cardíaca súbita es frecuente y puede estar provocada por arritmias ventriculares o isquemia. Tal como ocurre con la MHF, la MDF es una enfermedad heterogénea tanto desde el punto de vista clínico como genético. Se han descrito 6 loci genéticos diferentes: 1p1-1q1110, 1q32111, 2q31112, 3p22-25113, 9q13-q22114 y 10q21-23115, pero sólo la actina se ha identificado inequívocamente como causante de la MDF34. El mapeo cromosómico ha estado obstaculizado por los tamaños pequeños de las familias disponibles. El mapeo de los potenciales loci de la enfermedad en los cromosomas 1p1-1q1 y 3p25 también se complica por una penetrancia relacionada con la edad. El locus 1q32 tiene abundantes genes candidatos, que incluyen el factor de transcripción muscular MEF-2, la renina y la proteína que se une a la hélice-bucle-hélice del ADN MYF-4. El locus 1p1-1q1 contiene el gen codificador de la proteína de las gap junctions conexina 40, pero no se ha demostrado su relación causal. Olson et al34 han identificado recientemente 2 mutaciones con pérdida de sentido en la actina que causan la MCD; este hallazgo representa el primer gen autosómico relacionado con la enfermedad. Las mutaciones en la proteína del citoesqueleto desmina también pueden causar MCD35 y recientemente se han creado ratones transgénicos con una de las mutaciones116.
La desmina es un componente específico del músculo de la familia de filamentos intermediarios de las proteínas del citoesqueleto, y está expresada en los músculos estriado y liso. La proteína consiste en 470 aminoácidos (PM aproximado de 53 KDa) y está codificada por un único gen localizado en el cromosoma 2q33-q35. Contiene 9 exones. Es un componente fundamental del disco Z y contiene 3 dominios: una cabeza NH2-terminal no helicoidal, un dominio bastón helicoidal, que es crítico para el ensamblaje de los filamentos, y una cola COOH-terminal, cuya función sigue sin conocerse. En las fibras musculares estriadas maduras el entramado de los filamentos de desmina rodea los discos Z, los interconecta unos a los otros y une el aparato contráctil entero al citoesqueleto sarcomérico, los orgánulos citoplásmicos y el núcleo117.
Se han encontrado 7 mutaciones con pérdida de sentido en la región codificadora del gen de la desmina, 6 de ellas agrupadas en el bastón COOH-terminal118,119 y una en el dominio COOH de la cola35: esta última puede reflejar un dominio funcional bien diferenciado ya que sólo produjo enfermedad en el corazón. También se han descrito 2 deleciones adicionales que involucran los aminoácidos 7120 o 32121 en la región bastón. Las miocardiopatías relacionadas con la desmina son un grupo heterogéneo de enfermedades que se caracterizan por acúmulos intracitoplasmáticos de desmina122-125. Este desorden poco frecuente y a menudo familiar afecta al músculo esquelético y/o cardíaco y, a veces, está asociado con defectos en la musculatura lisa intestinal o vascular125,126. Las anormalidades del músculo esquelético se caracterizan por debilidad muscular y atrofia, mientras que en la enfermedad cardíaca se presenta a menudo un bloqueo de la conducción, arritmia e insuficiencia cardíaca restrictiva. Las miocardiopatías relacionadas con la desmina pueden consistir en una miocardiopatía dilatada o restrictiva con un pronóstico pobre después de un comienzo temprano en la edad adulta127.
Con la intención de investigar las relaciones entre estructura y función de las mutaciones de desmina y su capacidad para causar miopatías, se han generado 2 modelos de ratón usando reconstrucción génica mediante recombinación homóloga, y transgénesis. La generación de ratones sin desmina produjo animales viables y fértiles, lo que demuestra que la desmina no es necesaria para la formación del corazón o el alineamiento de las miofibrillas funcionantes128. Sin embargo, la desmina desempeña un papel esencial en el mantenimiento de la estructura y la integridad funcional de la miofibrilla, la fibra y el músculo entero, tal como se confirma por la presencia de anormalidades cardíacas, esqueléticas y del músculo liso poco después del nacimiento129,130. Las consecuencias fueron más severas en el corazón, que exhibió una degeneración progresiva y necrosis con calcificación extensa y fibrosis. Las alteraciones cardíacas incluyeron el de sarrollo de hipertrofia concéntrica en los cardiomiocitos, con inducción de expresión génica embrionaria. La dilatación ventricular y la función sistólica comprometida aparecieron más tardíamente131. No obstante, la relación que existe entre la mutación consistente en ausencia de desmina y la miopatía relacionada con la desmina no está clara, ya que la desmina total en la enfermedad cardíaca humana está significativamente aumentada.
La genética de la enfermedad en la población de pacientes humanos es confusa, ya que parece que al menos algunas de las mutaciones no se comportan como negativas dominantes. Para determinar si las mutaciones de desmina son una causa directa de las miopatías relacionadas con la desmina, se han generado ratones transgénicos116 que sobreexpresan la deleción de los 7 residuos de aminoácidos120. Los ratones que expresaban la desmina mutante presentaron agregados intrasarcoplásmicos aberrantes positivos para la desmina característicos de las miocardiopatías humanas relacionadas con la desmina (fig. 7). La morfología global del entramado de desmina estaba significativamente afectada. En los cardiomiocitos, el alineamiento de las miofibrillas se encontraba perturbado, y la morfología nuclear deformada. Tanto la contracción como la relajación de los miocitos del ventrículo izquierdo estaban significativamente comprometidas, así como también la función del órgano entero. Los ratones mutantes también desarrollaron una importante hipertrofia concéntrica. Así pues, este modelo demostró que las mutaciones actúan por una vía negativa dominante, y que la proteína mutante es la causa de la miocardiopatía resultante. Además, está pendiente de obtenerse nuevos datos sobre la progresión de la enfermedad y las relaciones entre causa y efecto que subyacen al proceso patogénico a partir de próximos análisis.

Fig. 7. Microscopia electrónica de agregados de desmina en un modelo de ratón de MCD. Secciones de corazones de controles de 8 semanas (A), sobreexpresión de la desmina del tipo original (B) y un animal transgénico que sobreexpresa una desmina encontrada en un paciente con MCD (C). Todas las muestras fueron obtenidas del ventrículo izquierdo. Tanto en (A) como en (B) la organización del sarcómero es normal con bandas I y A claramente distinguibles, con bandas M y bandas Z regularmente alineadas. (C) La acumulación de agregados electrodensos (*) rodeando el núcleo afecta la organización regular de los sarcómeros. El alineamiento de las bandas Z está alterado. A mayor magnificación (D), los agregados de desmina contienen material electrodenso (*) y filamentos intermediarios (flecha). Cabeza de flecha: filamentos de actina y miosina. N: núcleo.
El progreso futuro va a depender del descubrimiento de nuevas familias cuyos loci afectados puedan ser cuidadosamente mapeados para que los genes candidatos se puedan identificar y probar en modelos animales pero, en el momento actual, los mecanismos moleculares que conducen a la MDF autosómica dominante permanecen, en gran medida, sin conocerse. Además, la comprensión de las patologías que siguen un patrón de herencia autosómica dominante puede ser aún más difícil si tenemos en cuenta que al menos una parte de los casos de MDF es una enfermedad secundaria a otros procesos, como las miopatías esqueléticas, los defectos de conducción u otros desórdenes sistémicos. La ocurrencia de MDF también puede estar asociada a una herencia autosómica recesiva, herencia ligada al cromosoma X y herencia mitocondrial132-134. Aunque los mecanismos moleculares de algunas de estas patologías asociadas se han aclarado a partir de modelos experimentales y sistemas animales, que incluyen los modelos de ratones transgénicos135-137, el progreso que se ha obtenido a la hora de aplicar los resultados a los mecanismos que subyacen a la herencia autosómica dominante es extremadamente limitado.
La genética molecular de la MDF parece ser mucho más diversa que la de la MHF. Probablemente están involucradas otras estructuras celulares y orgánulos de los cardiomiocitos, tales como el citoesqueleto y la matriz extracelular. Pueden ser candidatos los genes para los componentes de la matriz, los factores de crecimiento, los receptores de las proteínas de señalización, los factores de transcripción, cada uno de ellos representando nuevos mecanismos posibles para causar la MDF autosómica dominante. De hecho, la sobreexpresión de un gran número de proteínas diferentes mediante una expresión específica en el corazón de animales transgénicos puede producir miocardiopatía dilatada7,15,138-142. La identificación futura del papel de estas proteínas y de sus dianas moleculares utilizando modelos animales transgénicos y de reconstrucción génica aportará una explicación para muchas de las miocardiopatías dilatadas heredadas y adquiridas.
Enfermedad cardíaca congénita
Aunque no es el objetivo de este trabajo de revisión, este tipo de enfermedad merece una mención por su prevalencia en la población pediátrica. La enfermedad congénita del corazón incluye varios desórdenes diferentes, clasificados como defectos miocárdicos, vasculares y arritmogénicos, y representa la principal causa de defectos en el nacimiento, que afecta a casi el 1% de los nacimientos a término143. Aunque muchas de las malformaciones cardíacas humanas están bien caracterizadas desde el punto de vista anatómico y fisiológico, las bases genéticas de estas anormalidades, que surgen durante la morfogénesis cardíaca, siguen sin ser bien conocidas. Con los recientes avances en genética y el desarrollo de tecnologías moleculares, especialmente el análisis de ligamiento y el clonaje posicional, los genes relacionados con las enfermedades han sido o están siendo identificados. Persisten las dificultades debido a que las proteínas involucradas en este síndrome son mayoritariamente desconocidas. Puesto que estos genes se han conservado durante la evolución en los vertebrados, muchos de los candidatos se identificaron por primera vez estudiando la regulación cardíaca y el desarrollo en sistemas animales simples que son más fáciles de utilizar para los análisis genéticos detallados, tales como las moscas, los nematodos y los peces cebra. Los candidatos son después validados en el ratón. La habilidad para manipular genéticamente los organismos simples y para realizar estudios mutagénicos de saturación ofrece una oportunidad para entender los potenciales agentes responsables del desarrollo cardíaco y su contribución a los defectos del corazón. Estos estudios han llevado a la identificación de numerosas proteínas, tanto estructurales como regulatorias, que pueden ejercer un papel fundamental en la función normal y anormal del corazón138,144-146.
DIRECCIONES FUTURAS
Durante los últimos 15 años, las técnicas de transgénesis y de reconstrucción génica han demostrado ser los métodos de elección para llevar a cabo experimentos genéticos en mamíferos. Las mejorías en las tecnologías deber proseguir para aportar métodos más afinados que permitan la exploración de las enfermedades cardiovasculares con modelos animales. Con respecto a la transgénesis, el objetivo más urgente tiene que ver con la capacidad para expresar transgenes de forma reversible; es decir, ser capaces de controlar la expresión de los transgenes en cualquier momento de la vida del animal. Una técnica que ofrece grandes promesas es el uso de tetraciclinas para controlar reversiblemente la expresión génica147,148. El paradigma tal como se describe es un sistema binario que requiere 2 transgenes: a) un promotor específico cardíaco que controla la secuencia transactivadora de la tetraciclina (tTA) acoplado a la proteína viriónica 16 (VP16) que actúa como factor de transcripción, y b) un promotor mínimo de citomegalovirus (CMV) acoplado a multímeros (5-7 copias) del operón de tetraciclina (tetO). Este promotor quimérico se liga entonces al transgén diana que está controlado condicionalmente. En presencia de tetraciclina, el tTA-VP16 se une al fármaco. La transcripción del gen indicador, que depende de que haya una actividad basal muy baja del promotor mínimo está «desconectada». Si la tetraciclina no está presente en el sistema, el tTA-VP16 se liga al tetO permitiendo que el transactivador VP16 aumente la expresión controlada por el promotor mínimo del CMV. La expresión del gen indicador puede así ser conectada y desconectada añadiendo o eliminando la tetraciclina. La transgénesis condicional puede ser una herramienta particularmente informativa para remodelar el sistema cardiovascular ya que una parte importante de la patología cardíaca humana está relacionada con mecanismos compensatorios inherentes a la naturaleza plástica del corazón. De manera alternativa, en un sistema tet diferente, las secuencias del operador tet son fusionadas a un promotor, y el tetR se convierte en una proteína transactivadora que activa la transcripción continuamente. En presencia de tetraciclina, el tTA no se puede ligar a las secuencias del operador tet, lo que produce una represión de la transcripción. La transgénesis condicional en el corazón ha sido demostrada149-152, aunque no está claro todavía si los sistemas actuales, en los que la expresión del transgén está controlada por la administración de tetraciclina u otros efectores, son lo suficientemente potentes como para controlar la gran variedad de proteínas necesaria para influenciar el contenido proteico sarcomérico. No cabe duda de que el sistema inducible ofrece el potencial de una herramienta muy poderosa para explorar las enfermedades cardiovasculares, ya que proporciona al investigador la oportunidad no sólo de observar si un transgén particular causa una enfermedad, sino también ver cómo la desconexión del transgén puede permitir la recuperación del corazón. Se pueden probar distintos tratamientos para determinar los métodos más efectivos para revertir la enfermedad.
Los avances en la reconstrucción génica van a estar centrados en conseguir la ablación génica de manera órgano-específica. Una de los principales inconvenientes de la reconstrucción génica tal y como se practica actualmente es la carencia de especificidad para dirigirse a un órgano. Es decir, la modificación génica es propagada a todas las células somáticas y, si el gen modificado es crítico para la función de otros órganos o sistemas musculares, puede complicar e incluso confundir a la hora de interpretar los efectos directos que ejerce sobre la función cardíaca. Se han intentado desarrollar modificaciones génicas que fuesen específicas para un órgano usando la familia enzimática de ADN recombinasa con el fin de superar esta dificultad, y se ha conseguido un progreso significativo con la Cre recombinasa. Este enfoque es aplastantemente eficaz. La ADN recombinasa Cre puede eliminar ADN en el genoma de los mamíferos con una gran eficiencia uniéndose a su secuencia diana: 34 pares de bases del ADN sustrato que reciben el nombre de loxP. Al flanquear la secuencia diana con los loxP en cada lado, y dirigiendo la expresión de la Cre específicamente al corazón mediante promotores cardíacos, debería ser posible restringir un evento de reconstrucción génica sólo al corazón al cruzar ratones que contengan el ADN flanqueado por los loxP con otras líneas que expresen la Cre en el compartimento cardíaco. Varios laboratorios han conseguido grandes progresos en la creación de líneas de ratones que cumplan estos requisitos, y acaban de ser publicados algunos trabajos en los que se describen resultados positivos de reconstrucción génica específica del corazón141,153,154. De hecho el sistema parece funcionar bastante bien; cuando los ratones son cruzados entre ellos, la endonucleasa se expresa específicamente en el corazón en aquellos animales que contienen los 2 loci. La enzima reconoce entonces el locus reconstruido mediante su unión a los lugares lox que están flanqueando la región y elimina el fragmento de ADN. El resultado es una ablación génica pero sólo en el corazón (o en el compartimiento cardíaco donde la endonucleasa se expresa específicamente). El procedimiento es bastante efectivo y abre incluso nuevas posibilidades de realizar estudios precisos de relación estructura-función y estudios de pérdida de función en el corazón.
Variaciones sobre estos temas, algunas de ellas todavía imprevisibles, nos van a permitir modular la función del corazón mediante intervenciones reversibles sobre los genes y las proteínas, de manera que se puedan determinar las bases moleculares de la función cardíaca normal y anormal. Éstas, a su vez, podrán ser usadas para explorar la fisiología cardíaca normal y anormal. En todos estos estudios, sean cuales sean los efectos que se obtengan, es importante tener presente que un ratón no es un hombre. La extensión de algunos de los modelos más valiosos a animales transgénicos grandes puede aportar, en primer lugar, una medida de la relevancia de las conclusiones derivadas de los estudios en modelos murinos y, en segundo lugar, modelos más adecuados para evaluar modalidades potenciales de tratamiento.
Sección patrocinada por el Laboratorio Dr. Esteve Correspondencia: J. Robbins. Division of Molecular Cardiovascular Biology Children's Hospital Research Foundation. Cincinnati, OH 45229-3039. EE.UU. Teléfono: 513-636-8098 Fax: 513-636-3852 Correo electrónico: jeff.robbins@chmcc.org