La hiperqueratosis palmoplantar aislada no ha sido descrita en asociación con cardiopatía; sin embargo, cuando esta queratodermia se acompaña de anormalidades en el pelo se han comunicado cardiopatías potencialmente severas. Queremos comunicar las alteraciones miocárdicas observadas en seis niños con hiperqueratosis palmoplantar con pelo lanoso pertenecientes a tres familias.
Palabras clave
Miocardiopatía
Ecocardiografía
Arritmias
INTRODUCCIÓN
Se denominan displasias del ectodermo (DE) a alteraciones de tipo hereditario en el desarrollo de estructuras 1 derivadas de esta placa germinativa. Las hiperqueratosis palmoplantares son DE que, además de afectar a piel y anexos, pueden comprometer otros tejidos de igual origen ectodérmico, como el sistema nervioso central 2, globos oculares, etc., respetando órganos y sistemas de diferente procedencia embrionaria.
En publicaciones de relativamente reciente aparición 3,4 se reseña ya la existencia de cardiopatía (mesodermo) asociada a un grupo de hiperqueratosis en las que, además de las alteraciones palmoplantares, se han observado lesiones en el pelo (pelo lanoso) que hacen de ésta una forma especial 5, tanto en el aspecto dermatológico como en el cardiológico, cuyas particularidades intentaremos, con la aportación de seis nuevos casos, dar a conocer en el presente estudio.
CASOS CLÍNICOS
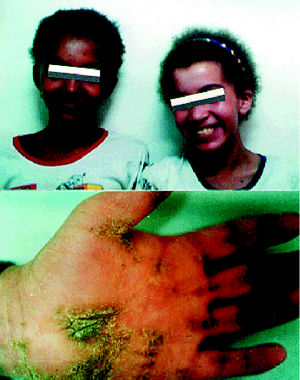
La población estudiada pertenece a la consulta de dermatología. Los tres primeros son casos nuevos, mientras que los restantes formaron parte de un estudio ya publicado 7 sobre hiperqueratosis palmoplantar familiar y pelo lanoso, alteraciones dermatológicas (fig. 1) que fueron criterios de inclusión.

Fig. 1. Pelo lanoso (superior) y queratodermia palmar (inferior).
Caso 1
Paciente de sexo femenino, de 10 años de edad, que ingresó por cansancio, disnea, edema pretibial, cuadro que un año antes había sido causa de internamiento en otro hospital con el diagnóstico de insuficiencia cardíaca congestiva.
En el examen físico se encontró hiperqueratosis palmoplantar y pelo fino, ensortijado, de aspecto lanoso desde el nacimiento. Las pestañas eran finas, largas y rizadas. Se observaba paquioniquia parcial en dedos de pies.
Los pulsos periféricos eran débiles y simétricos, la frecuencia cardíaca de 100 lat/min y la presión arterial de 90/60 mmHg.
Se apreciaba un choque de la punta desplazado hacia izquierda, un galope ventricular y soplo regurgitante mitral 2/6. Así mismo, se observaba hepatomegalia y edema pretibial.
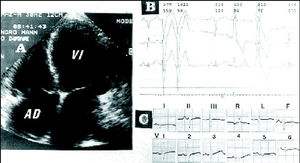
En la radiografía de tórax se observó cardiomegalia con redistribución venosa y derrame pleural derecho. La analítica sanguínea puso de manifiesto un ligero grado de anemia como único dato destacable. En el ECG se apreciaron ritmo sinusal, eje a la izquierda, trastornos de la conducción intraventricular, bajo voltaje y extrasistolia ventricular. La ecocardiografía-Doppler puso de manifiesto una marcada dilatación de cavidades cardíacas, sobre todo izquierdas (fig. 2A) con miocardio ventricular de espesor disminuido, brillante, con escasa contractilidad. En el pericardio grueso, se observó un derrame ligero. Mediante Doppler se detectó un llenado ventricular de tipo restrictivo y, además, insuficiencia mitral y tricuspídea moderadas.

Fig. 2. A: ecocardiografía en la que se aprecia gran dilatación de cavidades cardíacas (caso 1). B: Holter en el que se observa extrasistolia ventricular (caso 2). C: en el ECG se observa bajo voltaje, eje derecho (caso 3). AD: aurícula derecha; VI: ventrículo izquierdo.
El tratamiento con digitálicos, diuréticos, vasodilatadores y antiagregantes plaquetarios produjo una lenta disminución de la disnea y de los edemas, así como reducción de la hepatomegalia. El ecocardiograma de control hacia la cuarta semana demostraba aún dilatación de las cavidades, con leve pero apreciable mejoría de la contractilidad de los segmentos basales de las paredes ventriculares. Con un estado general mejorado y mientras se tramitaba el cateterismo y posiblemente la biopsia endomiocárdica, a realizarse en otra institución, la paciente sufrió un súbito deterioro hemodinámico con acentuada disnea, taquipnea, cianosis, náuseas y vómitos, estado del que, pese a las medidas adoptadas, no pudo ser rescatada falleciendo 24 h después. La oposición de los padres impidió realizar necropsia.
Caso 2
Niña de 12 años, hermana de la anterior, con pelo de tipo lanoso de color ligeramente claro, hiperqueratosis palmoplantar desde los 3-4 meses de edad. La paciente estaba asintomática y el examen físico era normal, con una presión arterial de 110/70 mmHg y una frecuencia cardíaca de 115 lat/min.
La analítica sanguínea era normal, así como la radiografía de tórax.
En el ECG se apreció ritmo sinusal, trastornos de la conducción intraventricular, bajo voltaje y ondas T negativas en D2, D3, aVF, V1 a V5. El Holter puso de manifiesto extrasístoles ventriculares apareadas (fig. 2B).
La ecocardiografia-Doppler puso de manifiesto una dilatación apical de ventrículo izquierdo con asinergia de ésta y de los segmentos apicales de las paredes inferior y lateral. No se observaron alteraciones valvulares y la función ventricular fue normal. Ante la ausencia de síntomas se decidió no instaurar tratamiento médico.
Caso 3
Varón de 17 años, hermano de las anteriores, que desde el nacimiento presentó pelo lanoso, fino, de color claro, con hiperqueratosis palmoplantar desde los 4 meses.
Acusaba disnea al esfuerzo. La auscultación cardíaca y pulmonar eran normales. En la analítica sanguínea se apreció una ligera anemia.
En el ECG se apreciaron ritmo sinusal, eje a la derecha y voltaje disminuido (fig. 2C), y la radiografía de tórax puso de manifiesto una cardiomegalia.
En la ecocardiografía-Doppler se evidenció una dilatación ventricular izquierda apical, con zonas de asinergia en los segmentos apicales de pared inferior. No se encontraron alteraciones valvulares. La fracción de eyección era del 47%.
El Holter puso de manifiesto extrasistolia ventricular frecuente, abundantes extrasístoles ventriculares apareadas y taquicardia ventricular no sostenida. El paciente se encuentra bajo tratamiento, permanece asintomático y acude a consulta de forma muy irregular.
Caso 4
Paciente femenina de 11 años de edad, con padres consanguíneos, con hiperqueratosis palmoplantar observada desde el año de edad, así como paquioniquia y paladar ojival, pelo lanoso claro desde el nacimiento.
No presentaba síntomas cardiovasculares, y el examen físico era normal, detectándose en la analítica solamente una ligera anemia. La radiografía de tórax era normal, y en el ECG se apreciaba ritmo sinusal, bajo voltaje, alteraciones de la conducción intraventricular, ondas T negativas en D3, aVF, V1 a V4 y extrasístoles ventriculares.
El Holter puso de manifiesto isquemia silente con arritmia ventricular 3 de Lown.
Se realizó una prueba de esfuerzo, que fue negativa para isquemia, evidenciándose en la fase de recuperación una mayor frecuencia y doble morfología en las extrasístoles ventriculares
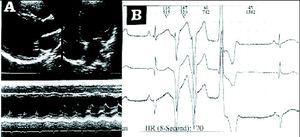
La ecocardiografía puso de manifiesto una dilatación ventricular izquierda con hipocinesia apical y segmentos apicales de paredes lateral e inferior (fig. 3A). La fracción de eyección era normal.

Fig. 3. A: ecocardiografía 2D que pone de manifiesto dilatación ventricular izquierda (caso 4). En modo M se aprecia hipocinesia de la pared inferior (cabezas de flecha). B: Holter en el que se observa taquicardia ventricular (caso 6).
Caso 5
Mujer de 12 años, hermana de la paciente anterior. Desde el nacimiento presentó pelo lanoso, fino y menos claro que el de su hermana. Presentaba hiperqueratosis palmoplantar desde los 12 meses de edad y paquioniquia parcial en pies.
No se observaron síntomas cardiovasculares, los pulsos y la presión arterial eran normales, así como la auscultación cardíaca y pulmonar.
La analítica sanguínea y la radiografía de tórax fueron normales. En el ECG se apreciaron bajo voltaje, ondas T negativas en D2, D3, aVF, V1 y V2, y extrasistolia ventricular.
La ecocardiografía-Doppler era normal, y en el Holter se observó arritmia ventricular 3 de Lown. Acude a consulta periódicamente, asintomática, sin tratamiento.
Caso 6
Varón de 14 años, primo hermano de las dos anteriores, con padres no consanguíneos. Presentaba pelo lanoso, claro y fino desde el nacimiento, las lesiones queratósicas en palmas y plantas aparecieron alrededor del año de edad. Se observaba paquioniquia parcial en pies.
Presentaba disnea al esfuerzo, y los pulsos y la presión arterial eran normales, así como la auscultación cardíaca y pulmonar.
En la analítica sanguínea no se apreciaron datos de interés.
La radiografía de tórax puso de manifiesto una cardiomegalia ligera sin alteraciones en la circulación pulmonar. La ecocardiografía-Doppler evidenció un leve crecimiento ventricular izquierdo. El ápex ventricular se encontraba dilatado y acinético, al igual que segmento contiguo de la pared inferior.
En el ECG se apreciaron desviación izquierda del eje y extrasístoles ventriculares.
El Holter puso de manifiesto extrasístoles ventriculares pareadas frecuentes y 2 episodios de taquicardia ventricular no sostenida (fig. 3B). El paciente se encuentra bajo tratamiento. DISCUSIÓN
Dentro de la variedad de las queratodermias palmoplantares, ninguna de ellas ha sido descrita en asociación con alteraciones cardíacas hasta hace poco tiempo, siendo las primeras publicaciones sobre el tema las de Protonotarios et al, quienes comunican su presencia en varios miembros de cuatro familias griegas. Posteriormente aparecen otros trabajos 3,4 que, junto a éste, confirman la relación entre la hiperqueratosis palmoplantar de tipo familiar con pelo lanoso y la miocardiopatía.
Las manifestaciones clínicas de cardiopatía abarcan trastornos en el ECG con alteraciones en el ritmo, voltaje, eje eléctrico y arritmias ventriculares. Además se observaron anormalidades en la contractilidad, dilatación y disfunción ventricular severa y muerte.
Aunque a primera vista parecen similares, las lesiones de nuestros pacientes son diferentes de las publicadas 3,4, ya que en ellas se observa hipertrofia ventricular derecha con una banda anómala corriendo paralela al septo ventricular, así como arritmia ventricular grave con muerte súbita en algunos casos, siendo muy poco trascendente la insuficiencia ventricular. Nuestros casos se caracterizaron por ausencia de estructuras paraseptales y de dilatación de cavidades derechas, estando mayormente comprometido el lado izquierdo con asinergia e insuficiencia ventricular. Las arritmias detectadas parecen ser menos agresivas.
Más recientemente 6 se han descrito tres casos fatales de miocardiopatía dilatada en niños pequeños con lesiones hiperqueratósicas y alopecia parcial o total. Estos nuevos casos difieren de los nuestros en las lesiones de pelo, y en cuanto a las lesiones cardíacas, la afectación predominante del ventrículo izquierdo con signos de miocardiopatía dilatada e insuficiencia cardíaca en los dos primeros y arritmia ventricular refractaria en el tercero, muy similares a nuestros casos.
Por el momento es difícil aseverar que se trate de una diferente respuesta cardíaca a una misma agresión o bien corresponda a dos tipos independientes de miocardiopatía, una que involucra al ventrículo derecho con arritmias graves como su manifestación principal, y otra con afectación del ventrículo izquierdo y signos y síntomas preferentemente de miocardiopatía dilatada e ICC.
Por limitaciones en los servicios de nuestro hospital no hemos podido avanzar en la búsqueda de la causa de esta enfermedad; sin embargo, por las detalladas investigaciones realizadas por otros autores 4-6 sabemos que no tiene un origen específico viral, infeccioso, autoinmune, coronario, etc. Al respecto, la propuesta de Hammill et al sobre un origen embrionario quedaría invalidada por el concepto de que las enfermedades embrionarias afectan sólo a tejidos de una misma placa. Sin embargo, se conoce en la actualidad 1 que estructuras complejas podrían ser el resultado de una interacción de placas, de forma que un tejido tendría origen en más de una hoja germinativa. Este criterio se ha comprobado experimentalmente al provocar, mediante la ablación de la placa neural (ectodermo) en embriones de pollo 7 alteraciones en la septación troncoconal (mesodermo), causando diversas malformaciones congénitas. Un punto en contra sería el hecho de que ni en trabajos anteriores ni en éste se encontraron cardiopatías congénitas asociadas; sin embargo, se ha observado, en los mismos modelos experimentales, que precediendo a estas malformaciones se producen cambios hemodinámicos con dilatación ventricular y fracción de acortamiento disminuida 8, provocados a su vez por una disfunción miocárdica primaria. Fuera del campo experimental, la interacción de ectodermo-mesodermo se observa en afecciones como la lentiginosis que, con frecuencia, se asocia a miocardiopatía hipertrófica 9.
La presencia familiar de esta alteración ectodérmica nos lleva a considerar un posible defecto genético. A pesar de que en la miocardiopatía dilatada familiar aún no se ha encontrado el gen responsable, en la cardiopatía que acompaña a la distrofia muscular de Duchenne 10 se ha identificado una anormalidad en el locus Xp21, responsable de la síntesis de una proteína estructural del miocito, hallazgo que apoyaría la hipótesis de un origen genético.
Aunque muchos aspectos de esta rara enfermedad permanecen en el plano especulativo, confiamos que investigaciones posteriores aporten mayor luz sobre el tema, pues la importancia de conocer esta extraña y grave asociación radica no solamente en el mal pronóstico que tiene la miocardiopatía dilatada en la población infantil 11, sino que, considerando las pautas de actuación clínica en estos pacientes, el tratamiento farmacológico 12 de la miocardiopatía dilatada debería empezar en la etapa asintomática, con la esperanza de retardar la fase de ICC y mejorar la supervivencia a largo plazo, actuación en la que nos encontramos y que esperamos sea motivo de una próxima comunicación. AGRADECIMIENTOS
Los autores dejan constancia de la invalorable asistencia de los Dres. Patricio Moncayo y Eduardo Guizado en la realización de los estudios Holter. AGRADECIMIENTOS
Durante la preparación del manuscrito tuvimos conocimiento del fallecimiento de la paciente 4. Había precisado hospitalización por insuficiencia cardíaca congestiva en dos ocasiones. A pesar de que su muerte se produjo en un área rural, se pudo realizar la autopsia, que puso de manifiesto, macroscópicamente, dilatación ventricular izquierda e, histológicamente, áreas de fibrosis intersticial.
Se denominan displasias del ectodermo (DE) a alteraciones de tipo hereditario en el desarrollo de estructuras 1 derivadas de esta placa germinativa. Las hiperqueratosis palmoplantares son DE que, además de afectar a piel y anexos, pueden comprometer otros tejidos de igual origen ectodérmico, como el sistema nervioso central 2, globos oculares, etc., respetando órganos y sistemas de diferente procedencia embrionaria.
En publicaciones de relativamente reciente aparición 3,4 se reseña ya la existencia de cardiopatía (mesodermo) asociada a un grupo de hiperqueratosis en las que, además de las alteraciones palmoplantares, se han observado lesiones en el pelo (pelo lanoso) que hacen de ésta una forma especial 5, tanto en el aspecto dermatológico como en el cardiológico, cuyas particularidades intentaremos, con la aportación de seis nuevos casos, dar a conocer en el presente estudio.
CASOS CLÍNICOS
La población estudiada pertenece a la consulta de dermatología. Los tres primeros son casos nuevos, mientras que los restantes formaron parte de un estudio ya publicado 7 sobre hiperqueratosis palmoplantar familiar y pelo lanoso, alteraciones dermatológicas (fig. 1) que fueron criterios de inclusión.

Fig. 1. Pelo lanoso (superior) y queratodermia palmar (inferior).
Caso 1
Paciente de sexo femenino, de 10 años de edad, que ingresó por cansancio, disnea, edema pretibial, cuadro que un año antes había sido causa de internamiento en otro hospital con el diagnóstico de insuficiencia cardíaca congestiva.
En el examen físico se encontró hiperqueratosis palmoplantar y pelo fino, ensortijado, de aspecto lanoso desde el nacimiento. Las pestañas eran finas, largas y rizadas. Se observaba paquioniquia parcial en dedos de pies.
Los pulsos periféricos eran débiles y simétricos, la frecuencia cardíaca de 100 lat/min y la presión arterial de 90/60 mmHg.
Se apreciaba un choque de la punta desplazado hacia izquierda, un galope ventricular y soplo regurgitante mitral 2/6. Así mismo, se observaba hepatomegalia y edema pretibial.
En la radiografía de tórax se observó cardiomegalia con redistribución venosa y derrame pleural derecho. La analítica sanguínea puso de manifiesto un ligero grado de anemia como único dato destacable. En el ECG se apreciaron ritmo sinusal, eje a la izquierda, trastornos de la conducción intraventricular, bajo voltaje y extrasistolia ventricular. La ecocardiografía-Doppler puso de manifiesto una marcada dilatación de cavidades cardíacas, sobre todo izquierdas (fig. 2A) con miocardio ventricular de espesor disminuido, brillante, con escasa contractilidad. En el pericardio grueso, se observó un derrame ligero. Mediante Doppler se detectó un llenado ventricular de tipo restrictivo y, además, insuficiencia mitral y tricuspídea moderadas.

Fig. 2. A: ecocardiografía en la que se aprecia gran dilatación de cavidades cardíacas (caso 1). B: Holter en el que se observa extrasistolia ventricular (caso 2). C: en el ECG se observa bajo voltaje, eje derecho (caso 3). AD: aurícula derecha; VI: ventrículo izquierdo.
El tratamiento con digitálicos, diuréticos, vasodilatadores y antiagregantes plaquetarios produjo una lenta disminución de la disnea y de los edemas, así como reducción de la hepatomegalia. El ecocardiograma de control hacia la cuarta semana demostraba aún dilatación de las cavidades, con leve pero apreciable mejoría de la contractilidad de los segmentos basales de las paredes ventriculares. Con un estado general mejorado y mientras se tramitaba el cateterismo y posiblemente la biopsia endomiocárdica, a realizarse en otra institución, la paciente sufrió un súbito deterioro hemodinámico con acentuada disnea, taquipnea, cianosis, náuseas y vómitos, estado del que, pese a las medidas adoptadas, no pudo ser rescatada falleciendo 24 h después. La oposición de los padres impidió realizar necropsia.
Caso 2
Niña de 12 años, hermana de la anterior, con pelo de tipo lanoso de color ligeramente claro, hiperqueratosis palmoplantar desde los 3-4 meses de edad. La paciente estaba asintomática y el examen físico era normal, con una presión arterial de 110/70 mmHg y una frecuencia cardíaca de 115 lat/min.
La analítica sanguínea era normal, así como la radiografía de tórax.
En el ECG se apreció ritmo sinusal, trastornos de la conducción intraventricular, bajo voltaje y ondas T negativas en D2, D3, aVF, V1 a V5. El Holter puso de manifiesto extrasístoles ventriculares apareadas (fig. 2B).
La ecocardiografia-Doppler puso de manifiesto una dilatación apical de ventrículo izquierdo con asinergia de ésta y de los segmentos apicales de las paredes inferior y lateral. No se observaron alteraciones valvulares y la función ventricular fue normal. Ante la ausencia de síntomas se decidió no instaurar tratamiento médico.
Caso 3
Varón de 17 años, hermano de las anteriores, que desde el nacimiento presentó pelo lanoso, fino, de color claro, con hiperqueratosis palmoplantar desde los 4 meses.
Acusaba disnea al esfuerzo. La auscultación cardíaca y pulmonar eran normales. En la analítica sanguínea se apreció una ligera anemia.
En el ECG se apreciaron ritmo sinusal, eje a la derecha y voltaje disminuido (fig. 2C), y la radiografía de tórax puso de manifiesto una cardiomegalia.
En la ecocardiografía-Doppler se evidenció una dilatación ventricular izquierda apical, con zonas de asinergia en los segmentos apicales de pared inferior. No se encontraron alteraciones valvulares. La fracción de eyección era del 47%.
El Holter puso de manifiesto extrasistolia ventricular frecuente, abundantes extrasístoles ventriculares apareadas y taquicardia ventricular no sostenida. El paciente se encuentra bajo tratamiento, permanece asintomático y acude a consulta de forma muy irregular.
Caso 4
Paciente femenina de 11 años de edad, con padres consanguíneos, con hiperqueratosis palmoplantar observada desde el año de edad, así como paquioniquia y paladar ojival, pelo lanoso claro desde el nacimiento.
No presentaba síntomas cardiovasculares, y el examen físico era normal, detectándose en la analítica solamente una ligera anemia. La radiografía de tórax era normal, y en el ECG se apreciaba ritmo sinusal, bajo voltaje, alteraciones de la conducción intraventricular, ondas T negativas en D3, aVF, V1 a V4 y extrasístoles ventriculares.
El Holter puso de manifiesto isquemia silente con arritmia ventricular 3 de Lown.
Se realizó una prueba de esfuerzo, que fue negativa para isquemia, evidenciándose en la fase de recuperación una mayor frecuencia y doble morfología en las extrasístoles ventriculares
La ecocardiografía puso de manifiesto una dilatación ventricular izquierda con hipocinesia apical y segmentos apicales de paredes lateral e inferior (fig. 3A). La fracción de eyección era normal.

Fig. 3. A: ecocardiografía 2D que pone de manifiesto dilatación ventricular izquierda (caso 4). En modo M se aprecia hipocinesia de la pared inferior (cabezas de flecha). B: Holter en el que se observa taquicardia ventricular (caso 6).
Caso 5
Mujer de 12 años, hermana de la paciente anterior. Desde el nacimiento presentó pelo lanoso, fino y menos claro que el de su hermana. Presentaba hiperqueratosis palmoplantar desde los 12 meses de edad y paquioniquia parcial en pies.
No se observaron síntomas cardiovasculares, los pulsos y la presión arterial eran normales, así como la auscultación cardíaca y pulmonar.
La analítica sanguínea y la radiografía de tórax fueron normales. En el ECG se apreciaron bajo voltaje, ondas T negativas en D2, D3, aVF, V1 y V2, y extrasistolia ventricular.
La ecocardiografía-Doppler era normal, y en el Holter se observó arritmia ventricular 3 de Lown. Acude a consulta periódicamente, asintomática, sin tratamiento.
Caso 6
Varón de 14 años, primo hermano de las dos anteriores, con padres no consanguíneos. Presentaba pelo lanoso, claro y fino desde el nacimiento, las lesiones queratósicas en palmas y plantas aparecieron alrededor del año de edad. Se observaba paquioniquia parcial en pies.
Presentaba disnea al esfuerzo, y los pulsos y la presión arterial eran normales, así como la auscultación cardíaca y pulmonar.
En la analítica sanguínea no se apreciaron datos de interés.
La radiografía de tórax puso de manifiesto una cardiomegalia ligera sin alteraciones en la circulación pulmonar. La ecocardiografía-Doppler evidenció un leve crecimiento ventricular izquierdo. El ápex ventricular se encontraba dilatado y acinético, al igual que segmento contiguo de la pared inferior.
En el ECG se apreciaron desviación izquierda del eje y extrasístoles ventriculares.
El Holter puso de manifiesto extrasístoles ventriculares pareadas frecuentes y 2 episodios de taquicardia ventricular no sostenida (fig. 3B). El paciente se encuentra bajo tratamiento. DISCUSIÓN
Dentro de la variedad de las queratodermias palmoplantares, ninguna de ellas ha sido descrita en asociación con alteraciones cardíacas hasta hace poco tiempo, siendo las primeras publicaciones sobre el tema las de Protonotarios et al, quienes comunican su presencia en varios miembros de cuatro familias griegas. Posteriormente aparecen otros trabajos 3,4 que, junto a éste, confirman la relación entre la hiperqueratosis palmoplantar de tipo familiar con pelo lanoso y la miocardiopatía.
Las manifestaciones clínicas de cardiopatía abarcan trastornos en el ECG con alteraciones en el ritmo, voltaje, eje eléctrico y arritmias ventriculares. Además se observaron anormalidades en la contractilidad, dilatación y disfunción ventricular severa y muerte.
Aunque a primera vista parecen similares, las lesiones de nuestros pacientes son diferentes de las publicadas 3,4, ya que en ellas se observa hipertrofia ventricular derecha con una banda anómala corriendo paralela al septo ventricular, así como arritmia ventricular grave con muerte súbita en algunos casos, siendo muy poco trascendente la insuficiencia ventricular. Nuestros casos se caracterizaron por ausencia de estructuras paraseptales y de dilatación de cavidades derechas, estando mayormente comprometido el lado izquierdo con asinergia e insuficiencia ventricular. Las arritmias detectadas parecen ser menos agresivas.
Más recientemente 6 se han descrito tres casos fatales de miocardiopatía dilatada en niños pequeños con lesiones hiperqueratósicas y alopecia parcial o total. Estos nuevos casos difieren de los nuestros en las lesiones de pelo, y en cuanto a las lesiones cardíacas, la afectación predominante del ventrículo izquierdo con signos de miocardiopatía dilatada e insuficiencia cardíaca en los dos primeros y arritmia ventricular refractaria en el tercero, muy similares a nuestros casos.
Por el momento es difícil aseverar que se trate de una diferente respuesta cardíaca a una misma agresión o bien corresponda a dos tipos independientes de miocardiopatía, una que involucra al ventrículo derecho con arritmias graves como su manifestación principal, y otra con afectación del ventrículo izquierdo y signos y síntomas preferentemente de miocardiopatía dilatada e ICC.
Por limitaciones en los servicios de nuestro hospital no hemos podido avanzar en la búsqueda de la causa de esta enfermedad; sin embargo, por las detalladas investigaciones realizadas por otros autores 4-6 sabemos que no tiene un origen específico viral, infeccioso, autoinmune, coronario, etc. Al respecto, la propuesta de Hammill et al sobre un origen embrionario quedaría invalidada por el concepto de que las enfermedades embrionarias afectan sólo a tejidos de una misma placa. Sin embargo, se conoce en la actualidad 1 que estructuras complejas podrían ser el resultado de una interacción de placas, de forma que un tejido tendría origen en más de una hoja germinativa. Este criterio se ha comprobado experimentalmente al provocar, mediante la ablación de la placa neural (ectodermo) en embriones de pollo 7 alteraciones en la septación troncoconal (mesodermo), causando diversas malformaciones congénitas. Un punto en contra sería el hecho de que ni en trabajos anteriores ni en éste se encontraron cardiopatías congénitas asociadas; sin embargo, se ha observado, en los mismos modelos experimentales, que precediendo a estas malformaciones se producen cambios hemodinámicos con dilatación ventricular y fracción de acortamiento disminuida 8, provocados a su vez por una disfunción miocárdica primaria. Fuera del campo experimental, la interacción de ectodermo-mesodermo se observa en afecciones como la lentiginosis que, con frecuencia, se asocia a miocardiopatía hipertrófica 9.
La presencia familiar de esta alteración ectodérmica nos lleva a considerar un posible defecto genético. A pesar de que en la miocardiopatía dilatada familiar aún no se ha encontrado el gen responsable, en la cardiopatía que acompaña a la distrofia muscular de Duchenne 10 se ha identificado una anormalidad en el locus Xp21, responsable de la síntesis de una proteína estructural del miocito, hallazgo que apoyaría la hipótesis de un origen genético.
Aunque muchos aspectos de esta rara enfermedad permanecen en el plano especulativo, confiamos que investigaciones posteriores aporten mayor luz sobre el tema, pues la importancia de conocer esta extraña y grave asociación radica no solamente en el mal pronóstico que tiene la miocardiopatía dilatada en la población infantil 11, sino que, considerando las pautas de actuación clínica en estos pacientes, el tratamiento farmacológico 12 de la miocardiopatía dilatada debería empezar en la etapa asintomática, con la esperanza de retardar la fase de ICC y mejorar la supervivencia a largo plazo, actuación en la que nos encontramos y que esperamos sea motivo de una próxima comunicación. AGRADECIMIENTOS
Los autores dejan constancia de la invalorable asistencia de los Dres. Patricio Moncayo y Eduardo Guizado en la realización de los estudios Holter. AGRADECIMIENTOS
Durante la preparación del manuscrito tuvimos conocimiento del fallecimiento de la paciente 4. Había precisado hospitalización por insuficiencia cardíaca congestiva en dos ocasiones. A pesar de que su muerte se produjo en un área rural, se pudo realizar la autopsia, que puso de manifiesto, macroscópicamente, dilatación ventricular izquierda e, histológicamente, áreas de fibrosis intersticial.
Bibliografía
[1]
The ectodermal dysplasias. Problems of classification and some newer syndromes. Arch Dermatol 1980; 116: 1295-1299.
[2]
Palmoplantar keratoderma and Charcot-Marie-Tooth disease. Arch Dermatol 1980; 116: 798-790.
[3]
Cardiac abnormalities in familial palmoplantar keratosis. Br Heart J 1986; 56: 321-326.
[4]
Woolly hair, palmoplantar keratoderma and cardiac abnormalities: report of a family. Arch Dermatol 1994; 130: 522-524.
[5]
Queratodermia epidermolítica asociada a pelo lanoso. Med Curt I L A 1995; 23: 279-284.
[6]
Cardiomyopathy with arrhythmias and ectodermal dysplasia: a previously unreported association. Am Heart J 1988; 115: 373-377.
[7]
Pathogenesis of persistent truncus arteriosus and dextroposed aorta in the chick embryo after neural crest ablation. Circulation 1987; 75: 255-264.
[8]
Hemodynamic changes Wall stresses and pressure gradients in neural crest-ablated chick embryos. En: Bockman DE, Kirby ML, editores. Embryonic origins of defective heart development. Nueva York: Ann NY Acad Sci 1990; 588: 305-313.
[9]
John Sutton MG, Tajik AJ, Giuliani ER, Gordon H, Su WPD. Hypertrophic obstructive cardiomyopathy and lentiginosis: a little known neural ectodermal syndrome. Am J Cardiol 1981; 47: 214-217.
[10]
X-linked dilated cardiomyopathy: molecular genetic evidence of linkage to the Duchenne muscular dystrophy (Dystrophin) gene at the Xp21 locus. Circulation 1993; 87: 1854-1865.
[11]
Idiopathic dilated cardiomyopathy in the young: clinical profile and natural history. J Am Coll Cardiol 1985: 6: 1126-1131.
[12]
Marcadores de la actuación clínica en pacientes con miocardiopatía dilatada. Rev Esp Cardiol 1996; 49: 682-692.