En la práctica clínica, en el contexto de la detección precoz de enfermedades y la prevención de enfermedades comunes de inicio en la edad adulta, uno de los problemas más habituales es la clasificación o toma de decisiones que se lleva a cabo mediante pruebas diagnósticas o pronósticas. Cuando se busca su optimización, es fundamental conocer su exactitud y su precisión.
Desde que se completó el Proyecto Genoma Humano, la combinación de proyectos de variación del genoma a gran escala, como el proyecto HapMap y el Proyecto 1.000 genomas, junto con el bajo coste de las plataformas de genotipado fiable y el rápido avance de las tecnologías de secuenciación del ADN, ha permitido estudios de asociación del genoma completo (genome-wide association studies [GWAS]) en cohortes grandes y estudios de secuenciación de exomas y genomas completos. En consecuencia, se ha producido un aumento exponencial de la abundancia de datos genotípicos específicos de cada individuo, lo que lleva a la era de la medicina personalizada o medicina de precisión basada en la genómica1.
Históricamente, las enfermedades genéticas se clasificaban en enfermedades de herencia mendeliana o simple, causadas por variaciones genéticas con un gran efecto, y enfermedades con una herencia compleja, causadas por la suma de variaciones genéticas de efecto reducido. Sin embargo, actualmente el riesgo general de cada individuo de contraer una enfermedad común probablemente se deba a una combinación de variantes genéticas frecuentes de bajo riesgo y variantes genéticas raras de alto riesgo2.
Los GWAS han priorizado la identificación de variantes genéticas asociadas con enfermedades o con rasgos (habitualmente polimorfismos de un solo nucleótido [SNP]), que son frecuentes en una población dada (p. ej., frecuencia de los alelos menores>1%). Hasta la fecha, los GWAS han identificado miles de loci asociados con varias características y afecciones humanas complejas, tales como las enfermedades cardiovasculares3. En particular, muchos de los loci previamente asociados con estas enfermedades humanas complejas destacan por múltiples SNP de bajo riesgo4.
Estos datos han proporcionado numerosos conocimientos sobre los genes y las vías que causan enfermedad, aunque más recientemente ha aumentado el interés por utilizar estos datos para predecir el riesgo de enfermedad5,6. En la última década, la medicina de precisión ha aparecido con fuerza con el objetivo de proporcionar atención sanitaria efectiva y adaptada a los pacientes en función de su dotación genética. La inclusión de puntuaciones de riesgo genético (GRS) con SNP asociados con enfermedad o fenotipo en la modelización del riesgo ha mejorado la exactitud en la predicción del riesgo individual7, tal como se explica en un artículo original publicado por Rincón et al. en Revista Española de Cardiología8.
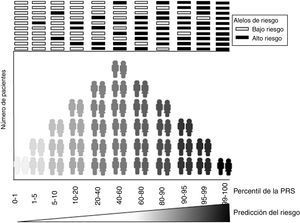
En la elaboración de los modelos de riesgo genético, es fundamental adquirir una capacidad predictiva precisa para identificar a los sujetos con riesgo (figura 1). Lo más habitual es que estos modelos se calculen como la suma ponderada del número de alelos de riesgo de un individuo, donde los alelos de riesgo y la magnitud de su efecto se definen en el GWAS6 previo. Así pues, la precisión de una GRS está determinada por la eficiencia de los GWAS previos para hallar variantes genéticas asociadas con enfermedades comunes. En otras palabras, cuanto más estables sean los cimientos del edificio -n este caso, las asociaciones genéticas descritas en los GWAS- más resistente será la construcción, es decir, más exacta será la estimación de la predicción del riesgo.
El rendimiento predictivo suele evaluarse mediante las curvas de características operativas del receptor (ROC), en las que la sensibilidad y la especificidad de las predicciones se clasifican en varios puntos de corte. En el caso simplista de predicción de la aparición de cualquier afección o enfermedad, la sensibilidad se da como la fracción de la razón de verdaderos positivos entre el total de pacientes enfermos. Cabe destacar que un verdadero positivo es cualquier paciente que padece la enfermedad y tiene un resultado positivo en el modelo de predicción clínica. La razón de verdaderos positivos es la probabilidad de clasificar correctamente a un paciente. La especificidad es la razón de verdaderos negativos entre el total de pacientes no enfermos. La especificidad es la probabilidad de clasificar correctamente a un sujeto sano, es decir, la probabilidad de que una persona sana dé un resultado negativo. Una curva ROC es una representación gráfica bidimensional en la que la razón de verdaderos positivos (sensibilidad) se representa en el eje vertical y la de falsos positivos (1 - especificidad), en el eje horizontal. Por lo tanto, un gráfico ROC de un modelo de predicción representa el equilibrio relativo entre verdaderos positivos y falsos negativos. El área bajo la curva ROC es la probabilidad de que el modelo examinado identifique correctamente el caso entre un caso y un control elegidos al azar. Los resultados del área bajo la curva ROC oscilan entre 0,5 (aleatorio) y 1 (100% de exactitud).
Las GRS o puntuaciones de riesgo poligénico (PRS), como muchos autores las denominan ahora, más que predecir la presencia o ausencia de una enfermedad, tienen por objetivo clasificar la población en distintos niveles de riesgo (figura 1). El umbral para considerar que una GRS es positiva depende del equilibrio entre el valor de riesgo marcado por sus propios valores límite, junto con otros factores de riesgo y las ventajas asociadas con una posible intervención terapéutica o en el estilo de vida.

Actualmente se cree que la genética de las formas no familiares de las enfermedades cardiacas comunes de inicio en la edad adulta se relaciona principalmente con una combinación de variantes frecuentes con efecto reducido distribuidas por todo el genoma y de variantes raras de efecto moderado situadas en genes conocidos por causar la forma familiar de la enfermedad. Se han descrito pruebas de ello en estudios de genómica exhaustivos y recientes, tales como un extenso GWAS sobre enfermedad coronaria9 y un estudio de secuenciación a gran escala de la diabetes mellitus tipo 210. El efecto de cada una de estas variantes frecuentes en un sujeto será demasiado pequeño para predecir el riesgo, pero la combinación de muchas de estas variantes frecuentes puede utilizarse para predecir eficazmente el riesgo, sobre todo si la predicción de dicho riesgo se realiza conjuntamente con factores de riesgo clásicos tales como los factores de riesgo clínico o determinadas exposiciones ambientales.
Una de las primeras publicaciones sobre la implementación de las GRS en las enfermedades cardiovasculares fue el estudio de Morrison et al.11, donde el uso de una puntuación de 11 polimorfismos para predecir el riesgo de enfermedad coronaria no mejoró la capacidad predictiva de los factores de riesgo clásicos. Desde entonces, se han publicado y validado muchas PRS, que son especialmente eficaces en grupos de pacientes con fenotipo muy específico. Algunos ejemplos son las PRS de la enfermedad coronaria, cuyo objetivo es individualizar la decisión de iniciar el tratamiento de por vida con estatinas12, o las PRS para mejorar la capacidad de predicción de los pacientes clasificados con un riesgo intermedio de enfermedad cardiovascular según la escala de Framingham13.
La GRS aplicada a adultos jóvenes para predecir los episodios recurrentes tras el infarto de miocardio, tal como describen Rincón et al.8, debería validarse en una muestra más amplia, ya que los resultados positivos se observan principalmente en pacientes jóvenes sin diabetes, pero la observación se basa en un número reducido de pacientes. Estudios como este abren la puerta a la implementación de las PRS en los modelos de predicción clínica, aunque estos siempre tienen que validarse y basarse en numerosos datos.
Por último, no hay que olvidar la incertidumbre en la estimación de la magnitud de efecto asociada con cada una de las variantes frecuentes incluidas en una puntuación genética, cuando se utiliza la PRS para estimar el riesgo en otras poblaciones distintas de las estudiadas en el GWAS. Puesto que la mayor parte de los GWAS se ejecutaron en poblaciones de origen europeo y es bien conocida la diversidad genética entre las poblaciones de origen distinto, hay que tener especial cuidado a la hora de generalizar la aplicabilidad de las PRS a todas las poblaciones del mundo. Los cálculos no son transferibles entre poblaciones, y la PRS se aplica a un paciente de origen geográfico determinado y con una dotación genética característica, pero con los mismos derechos a la atención sanitaria14.
Aunque el número de estudios con estimaciones del riesgo poligénico ha crecido exponencialmente en los últimos 5 años, deberían llevarse a cabo estudios a gran escala para demostrar la utilidad de la estimación del riesgo poligénico no solo en el ámbito cardiovascular, sino también en otras áreas de la salud humana. En esta línea, la iniciativa europea «1 millón de genomas», cuyo objetivo es conseguir en 2022 este número de genomas secuenciados vinculados a datos clínicos, con España como estado miembro signatario, es quizás el proyecto más prometedor15. Los datos de todo el genoma a esta escala tienen potencial para avanzar rápidamente en la medicina de precisión y las estimaciones de predicción del riesgo.
FINANCIACIÓNEste estudio fue financiado en parte por el Plan Estatal de I+D+i 2013-2016, Subdirección General de Evaluación y Fomento de la Investigación (ISCIII-SGEFI) del Instituto de Salud Carlos III (ISCIII) y el Fondo Europeo de Desarrollo Regional (FEDER) (subvenciones número PI16/00903, CB16/11/00226, CB06/07/0088).
CONFLICTO DE INTERESESNinguno.