Los descubrimientos de los aspectos moleculares del funcionamiento celular están cambiando los conceptos de salud y enfermedad. Todas las áreas de la medicina, incluyendo la cardiología, se enriquecen con pruebas diagnósticas para determinar la predisposición y realizar la detección de alteraciones moleculares. Esta revisión acerca de la genética y de los aspectos moleculares en cardiología se escribe en el centenario del redescubrimiento de los principios de Mendel y en el momento en que se anuncia la secuenciación del genoma humano. El artículo comienza con consideraciones acerca de la constitución pluricelular del cuerpo humano y de los principios de la genética y sus bases moleculares, incluyendo una breve descripción de los métodos de mapeo genético. A continuación, se hace una reseña histórica sobre la genética médica, la medicina molecular y el Proyecto del Genoma Humano. Finalmente se realiza una exposición sobre el espectro de enfermedades genéticas, utilizando ejemplos de afecciones cardiovasculares.
Palabras clave
Enfermedades cardiovasculares
Gen, herencia
Genética médica
Medicina molecular
Proyecto del Genoma Humano
CONSTITUCIÓN PLURICELULAR Y PROCESOS DE DIVISIÓN CELULAR
La información genética almacenada en una célula es requerida en dos procesos biológicos universales: la transmisión de características morfológicas y funcionales similares a sus descendientes y la producción de biomoléculas complejas como el ácido ribonucleico (ARN) y las proteínas, los cuales permiten la construcción de la célula, su funcionamiento y el establecimiento de relaciones con las células vecinas y con el entorno ambiental que la rodea.
Por lo que se refiere a las posibilidades de la reproducción, la cual asegura la subsistencia de la especie, los organismos pluricelulares (metazoarios) como la especie humana están constituidos por dos tipos de células: los gametos (óvulo y espermatozoide), células encargadas de la generación de nuevas criaturas a partir de la reproducción sexual, y las células somáticas, o el resto de células que constituyen el organismo y cuya función más importante, en este contexto, es asegurar el éxito de la participación de los gametos en la reproducción. Las células que constituyen los tejidos del corazón y del sistema vascular pertenecen a este último grupo. Cualquier alteración que afecte la forma o la función del sistema cardiovascular en estadios previos a la reproducción del sujeto afectado limitaría sus posibilidades para establecer una descendencia y perpetuar su información genética. Las células somáticas se reproducen mediante un proceso simple de división, el cual ocurre principalmente durante las etapas de desarrollo del individuo y con excepción de algunos tejidos (como el sistema hematopoyético), se detiene casi por completo en el resto del cuerpo después del nacimiento (p. ej., en el miocardio). Estos dos procesos de división celular que ocurren en el cuerpo humano se considerarán a continuación.
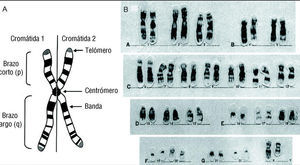
La transmisión de la información genética a las células descendientes tiene algunos elementos y particularidades. Se debe considerar la existencia de una unidad mínima de transmisión de la información hereditaria y de su comportamiento. Este elemento recibe el nombre de gen y está encargado de transmitir una característica particular a la célula hija 1. El conjunto y las interacciones de todas las características transmitidas por múltiples genes determinarán al individuo. Los genes, que se describirán más adelante con más detalle, están inmersos en el material genético que, en el caso de nuestra especie, es el ácido desoxirribonucleico (ADN) 1. La carga de material genético presente en las células se denomina genoma. Los gametos poseen una sola copia de genoma y por eso se denominan células haploides, mientras que las células somáticas poseen dos copias genómicas y se denominan diploides. Los genes están albergados en enormes estructuras cromáticas del núcleo celular, denominadas cromosomas. Durante la profase y metafase mitóticas, que se describirán posteriormente, los cromosomas se individualizan dentro del núcleo y originan estructuras de forma definida que pueden ser analizadas. En nuestra especie, se pueden distinguir dos grupos de cromosomas: los que determinan el sexo de un sujeto (cromosomas sexuales X e Y) y 22 cromosomas adicionales denominados autosomas 1. El cariotipo es un estudio de laboratorio en el que se analizan la constitución y estructura de los cromosomas de un sujeto, a partir de un cultivo de linfocitos extraídos de su sangre periférica 2. Este estudio revelará la naturaleza diploide de las células somáticas y demostrará que cada cromosoma está representado por un par, con excepción de los varones, en los que se observará una copia de cromosoma X y otra del Y 1. Los componentes de cada par se denominan cromosomas homólogos. Este análisis es crucial para el estudio de algunas enfermedades hereditarias que afectan el corazón, ya sea por alteración del número de cromosomas (síndrome de Down, producido principalmente por la presencia de tres copias -trisomía- del cromosoma 21 y que presenta diversas malformaciones cardíacas) 3, o por alteración de la estructura (síndrome cri du chat, producido por pérdida de material genético -deleción- del brazo corto del cromosoma 5, que se acompaña particularmente de defectos del tabique interventricular) 3. La figura 1A representa la anatomía de un cromosoma y en la figura 1B se ilustra el cariotipo de un sujeto normal, en el cual se agrupan los cromosomas en orden de tamaño y forma para ser estudiados.

Fig. 1. A: anatomía básica de un cromosoma metacéntrico. En este caso, se ilustra un cromosoma replicado con dos cromátidas que se segregarán a las células hijas durante la división celular. B: cariotipo de un varón normal. El cariotipo es la disposición ordenada de los 23 pares de cromosomas humanos de una célula en división mitótica, en grupos determinados por el tamaño y la morfología de los cromosomas. (Cortesía de la QFB Itzel Calleja, Facultad de Medicina, UANL.)
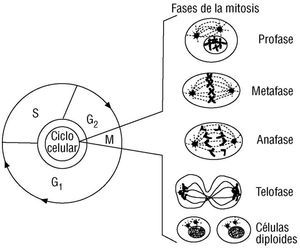
El proceso que gobierna la división de las células somáticas se denomina mitosis (fig. 2), mientras que el que gobierna la división de los precursores de los gametos maduros se denomina meiosis (fig. 3) 1. La mitosis se inicia desde el momento mismo de la fecundación, es decir, desde la formación del cigoto; mientras que la meiosis ocurre más tardíamente, después de la formación de las gónadas en el embrión del nuevo individuo. Durante la mitosis, cada cromosoma se replica a sí mismo a lo largo de la interfase, generando las dos mitades del cromosoma denominadas cromátidas (fig. 1A). Durante la profase los cromosomas se condensan gradualmente para tomar su forma característica. Durante la metafase los cromosomas se agrupan a lo largo de una línea imaginaria vertical que divide el núcleo en dos mitades, con cada cromátida orientándose hacia uno de los dos polos nucleares resultantes. Finalmente, durante las etapas de anafase y telofase cada cromátida migra y se establece en cada una de las mitades nucleares, al tiempo que la línea imaginaria mencionada sirve de guía para la separación y reconstitución de las membranas nuclear y celular de dos nuevas células. Las células generadas entran en el período de interfase 1. Los sucesos de replicación durante la interfase se explicarán después de la meiosis.

Fig. 2. Ciclo celular y mitosis. El ciclo celular se inicia cuando la célula se prepara para la división celular y concluye con la generación de dos células hijas. En la fase G1 la célula verifica las condiciones metabólicas óptimas para el proceso de división. Durante la fase S el ADN cromosómico se replica. Una vez ha concluido la síntesis de ADN, se inicia la fase G2 y la célula nuevamente verifica las condiciones metabólicas requeridas para iniciar el proceso de división, el cual ocurre durante la fase M o mitosis. La mitosis es el proceso de división celular que gobierna a las células somáticas. En este proceso, el material cromosómico ya replicado se condensa gradualmente en los cromosomas como el que se ilustra en la figura 1A. Estos cromosomas se reúnen en el centro del núcleo en división y los centrómeros se separan verticalmente. Cada nuevo cromosoma (cromátida) inicia su migración hacia los polos opuestos del núcleo. En la fase final, las membranas nuclear y plasmática se dividen y reconstituyen para generar dos células hijas diploides. Este tipo de división se requiere para los procesos de crecimiento, morfogénesis, desarrollo y regeneración de tejidos en el cuerpo humano. Al final del proceso se conservan los genomas diploides en las células hijas.

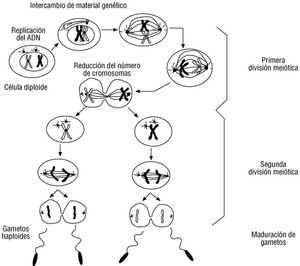
Fig. 3. Meiosis. La meiosis es el proceso de división celular que gobierna a las células germinales. Este proceso consta de dos etapas de división celular consecutivas, denominadas meiosis I y meiosis II. En la meiosis I ocurren dos fenómenos cruciales: la recombinación de segmentos cromosómicos, que es la principal fuente de variación genética en las especies que se reproducen sexualmente, y la reducción del número de cromosomas para constituir un óvulo o un espermatozoide con genoma haploide. Durante la fecundación, el cigoto reunirá los dos genomas de las células participantes para restablecer el genoma diploide y comenzar el crecimiento y desarrollo de un nuevo ser.
La meiosis es más compleja. Este proceso consta de dos pasos de división celular consecutivos denominados meiosis I y meiosis II, en los cuales se reduce el genoma de la célula primordio de un gameto hasta lograr una constitución haploide en el producto final 1. También ocurren otros fenómenos cruciales desde el punto de vista hereditario que deben ser considerados. Durante la interfase, al inicio de la meiosis I (fig. 3), cada cromosoma se replica y da origen a las cromátidas correspondientes. A continuación los cromosomas formados se compactan durante la profase I pero, a diferencia de la mitosis, cada cromosoma se alinea en el núcleo con su par homólogo. La alineación resultante genera una estructura denominada bivalente, que permite el intercambio de material genético entre las cuatro cromátidas participantes. Este complejo proceso implica la reunión estrecha de dos cromátidas en un punto específico, denominado quiasma, la rotura de los cromosomas, la reunión de dos cromátidas de diferente origen y la ligazón de los extremos resultantes en dicho punto, lo que se denomina recombinación cruzada. Los cromosomas originales, de los cuales una copia era heredada del espermatozoide (cromosoma paterno) y la otra provenía del óvulo (cromosoma materno), ya no son los mismos después de ocurrir este proceso y ésta es una de las bases de la variación genética entre hermanos y, más ampliamente, de las variaciones genéticas en las poblaciones humanas 1. También es una de las bases de los estudios de ligamiento (que se discutirán posteriormente). Durante la metafase I, los quiasmas desaparecen y cada par de cromosomas homólogos, aún constituidos por dos cromátidas, se reúne a lo largo de la línea vertical que divide el núcleo e inician su migración a cada polo. En las etapas de anafase y telofase I se concluye la división celular y el resultado son dos células que ahora poseen genomas haploides, replicados y recombinados. La interfase de la meiosis II es muy corta o puede ser inexistente, y el material genético no se replica nuevamente. En las etapas profase y metafase II los cromosomas se alinean nuevamente en el eje divisorio vertical y las cromátidas de cada uno se separan y migran a los polos resultantes para constituir el gameto resultante al final de las anafase y telofase II: un óvulo o un espermatozoide maduro 1. La fecundación entonces reunirá a los genomas haploides de estos dos gametos para restablecer el genoma diploide e iniciar el desarrollo y el crecimiento del nuevo ser 1.
PRINCIPIOS DE LA HERENCIA
Con las consideraciones previas, se pueden introducir algunos conceptos sobre la herencia. El momento en el que se escribe este artículo se celebra un centenario muy importante para la biología y la medicina: el del curioso redescubrimiento de los principios formulados por Gregor Mendel en 1866 4, que fue realizado de forma independiente por tres biólogos y botánicos: Hugo de Vries en Holanda, Carl E. Correns en Alemania y Erich Tschermak von Seysenegg en Austria. Las observaciones de Mendel y de los investigadores mencionados se realizaron gracias a la identificación de características morfológicas particulares en plantas progenitoras y sus descendientes. A esta característica reconocible se le denomina fenotipo, y a la información genética que la determina se le denomina genotipo 1. El genotipo es una secuencia de ADN determinada que constituye un gen y cuyo producto expresado determina el fenotipo 1. Los genotipos pueden tener formas alternas, denominadas alelos, las cuales resultan de mutaciones de la versión original del gen. Algunas mutaciones no tienen efecto patológico, como los polimorfismos, que se explicarán más adelante, y otras variantes son las causantes de las enfermedades monogénicas. El Catálogo de la Herencia Mendeliana del Hombre, una importante compilación dirigida por el Dr. Victor A. McKusick, agrupa todos los genes humanos descritos en un banco de datos disponible en Internet (www.ncbi.nlm.nih.gov/omim) 5. Cada afección mendeliana humana tiene un número en este catálogo (MIM), el cual será utilizado en este artículo para referir al lector que desee información genética y clínica adicional sobre el cuadro particular mencionado. Para explicar las formas alélicas de un gen, se puede tomar como ejemplo al gen de la distrofina, localizado en el brazo corto del cromosoma X y que codifica para una gran proteína constituyente de las fibras musculares esqueléticas, lisas y cardíacas 6. Este gen puede presentar variantes genotípicas responsables de, por lo menos, cuatro fenotipos, uno normal y tres patológicos: las distrofias musculares de Duchenne y de Becker [MIM 310200], que afectan a las fibras musculares esqueléticas de forma generalizada y que también suelen afectar las fibras cardíacas, y la miocardiopatía dilatada ligada al X, que afecta particularmente al corazón 7.
El primer principio de Mendel, denominado segregación independiente, establece que cada gameto recibe uno de los dos alelos posibles (uno materno y otro paterno), sin que medie ningún mecanismo de selección en la gónada 1. Con este principio se entenderá en el siguiente ejemplo que si un varón porta un alelo silvestre o normal del gen de la fibrilina 1 y otro alelo mutado, durante la formación de los espermatozoides, la mitad portará el alelo normal, y la restante, el alelo mutado. Además, los espermatozoides estarán distribuidos al azar y todos tendrán la misma posibilidad de participar en la fecundación. Los alelos mutados del gen fibrilina 1 producen el síndrome de Marfan [MIM 154700], una enfermedad mendeliana que afecta a la aorta y al corazón, entre otros órganos 8. Los descendientes que reciben el alelo mutado sufrirán la enfermedad. Como en este caso la herencia de un solo alelo mutado es suficiente para que la enfermedad se manifieste, se dice que este alelo tiene un fenotipo dominante. En el caso de la ataxia de Friedreich [MIM 229300], que puede acompañarse de miocardiopatía, el sujeto afectado recibe los dos alelos mutados de cada uno de sus padres. La ausencia del alelo silves-tre que tendría un comportamiento dominante, permite que la enfermedad se exprese y se establece que esta enfermedad tiene un fenotipo recesivo 9. Como se observa con estos ejemplos, la clínica ofrece la posibilidad de estudiar los efectos de la herencia mendeliana al permitir establecer fenotipos que pueden reconocerse en el paciente y en los miembros de su árbol genealógico, una práctica que cada vez gana más importancia cuando el profesor de semiología examina las historias clínicas de sus discípulos.
El segundo principio, denominado distribución independiente, establece que los alelos de dos genes, aun cuando puedan estar localizados en el mismo cromosoma, se pueden separar durante la meiosis I y repartirse de forma independiente en los gametos resultantes 1. Por ejemplo, los genes responsables del síndrome de DiGeorge [MIM 188400], caracterizado por malformaciones cardíacas, inmunodeficiencia celular debida a hipoplasia tímica e hipoplasia de tiroides y paratiroides, están en la banda q11 del cromosoma 22. En este mismo cromosoma reside el gen CYP2D [MIM 124030], que codifica para un producto de la fracción microsomal p450 involucrado con el metabolismo de la debrisoquina y el propranolol, medicamentos betabloqueadores utilizados en el control de la hipertensión arterial. El locus de este gen se localiza en la banda q13 del mismo cromosoma 22. Las variantes alélicas del gen CYP2D pueden determinar sujetos que son normosensibles, hipersensibles y resistentes a este tipo de medicamentos. Según esta definición fisiológica del fenotipo, imaginemos un árbol genealógico en el que se está heredando el síndrome de DiGeorge y en el cual además hay sujetos normosensibles o hipersensibles a los betabloqueadores mencionados. La segregación de los dos fenotipos a través de las generaciones presentará proporciones similares de sujetos sin malformación normosensibles, de sujetos sin malformación hipersensibles, de sujetos malformados normosensibles y de sujetos malformados hipersensibles. Con este resultado se corroborará que la segregación de la malformación no determina el tipo de sensibilidad al betabloqueador y que cada gen es una entidad independiente.
Algunas veces este principio es quebrantado. Examínese el siguiente caso: el gen de la enzima 21-hidroxilasa es vecino del gen del antígeno leucocitario humano-B (HLA-B) en el brazo corto del cromosoma 6. El primer gen puede estar representado por un alelo silvestre (HSR) y un alelo mutado (hsr) que puede producir hiperplasia suprarrenal congénita [MIM 201910] en sujetos homocigotos (hsr/hsr), una de cuyas variedades puede acompañarse de hiperpotasemia y arritmia 10. El gen vecino puede producir varias formas de la misma proteína que no tienen ninguna implicación patológica (sólo poseen características antigénicas diferentes) y determinan lo que se denomina un sistema polimórfico (un polimorfismo es una variante alélica sin repercusión patológica que está presente en, por lo menos, un 10% de una población estudiada; otros buenos ejemplos de sistemas polimórficos son los grupos sanguíneos) 1. Supóngase que un individuo portador de la mutación para hiperplasia suprarrenal congénita puede tener dos tipos de cromosomas 6: el primero porta los alelos hsr y HLA-Bw47, y el homólogo correspondiente tiene los alelos HSR y HLA-B8. Al aplicar el principio de la distribución independiente, las gónadas de este sujeto generarían proporciones similares de gametos con los siguientes genotipos: hsr/HLA-Bw47, hsr/HLA-B8, HSR/HLA-Bw47 y HSR/HLA-B8. Ocurre algunas veces que estas proporciones no resultan de acuerdo con lo planteado por este principio, debido a un fenómeno denominado ligamiento de genes 11.
MAPAS GENÉTICOS
El ejemplo anterior tiene la particularidad de que el principio de distribución independiente es desobedecido. En el sujeto referido, las proporciones de distribución encontradas en los gametos no resultarán de acuerdo con las calculadas, debido a que la vecindad de los genes afecta la posibilidad de formación de un quiasma en la meiosis I que pueda separar a los dos genes. El resultado es que, en la mayoría de los gametos, los alelos originales se reparten sin haberse cambiado de cromátida. Este fenómeno se denomina ligamiento y el estudio de esta eventualidad espacial ha permitido la ubicación de genes en los cromosomas y determinar las distancias genéticas que separan a éstos en un cromosoma. Éstos son los principios básicos del mapeo de genes. Es muy importante mencionar que biólogos matemáticos como Thomas H. Morgan lograron realizar mapas genéticos y asignación de distancias entre un gen y otro con sólo consideraciones matemáticas 12,13. Estos biólogos tomaron como modelo de estudio la mosca de la fruta (Drosophila melanogaster). Después de realizar varios cruces sucesivos y generar abundantes árboles genealógicos constituidos por varias generaciones de individuos, eran capaces de analizar simultáneamente determinados fenotipos mutantes en la descendencia. Estos experimentos se realizaron a principios del siglo xx, antes de que se supiera que el material genético de los cromosomas era una doble cadena lineal de ADN en la que se ubicaban los genes y en la cual las distancias que separan a dos genes pueden establecerse físicamente en términos de pares de bases.
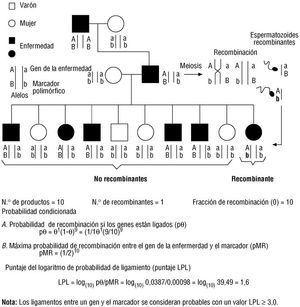
El hallazgo de árboles genealógicos útiles y abundantes y la realización de estudios de ligamiento no son tareas sencillas en la especie humana, debido a la imposibilidad de seleccionar experimentalmente a sujetos para la reproducción y de observar todos los productos que se formarían con los gametos generados en un varón y una mujer, así como por el tiempo que representaría estudiar a varias generaciones para analizar los genotipos y fenotipos resultantes. De acuerdo con estas consideraciones, los genetistas han ideado métodos para el estudio de la transmisión de un fenotipo (estudios de segregación en las familias, de concordancia en gemelos, el método del par de hermanos - sib pair, en inglés-, etc.) y herramientas matemáticas que analizan el ligamiento entre un gen y los marcadores vecinos, considerando una condicionante probabilística que haya ocurrido recombinación en la meiosis 14,15. El método más utilizado es la puntuación del logaritmo de la probabilidad de ligamiento o LOD score, en inglés (fig. 4) 16.

Fig. 4. Estudio de ligamiento y puntuación del logaritmo de la probabilidad de ligamiento (LPL) o LOD score. Se representa un árbol genealógico en el que se está segregando una enfermedad monogénica autosómica dominante. Debajo de cada individuo se representa el orden de alelos de un gen de una enfermedad y de un marcador genético polimórfico, sospechosamente ligados en un segmento cromosómico particular y discriminados por su ubicación en los cromosomas homólogos materno y paterno. Para el análisis de ligamiento es importante determinar el número de sujetos que demuestran el fenómeno de recombinación (intercambio genético que genera cromosomas con un nuevo orden de alelos, como se ilustra en la porción superior derecha de la figura). El recuento de sujetos recombinantes en los diferentes árboles genealógicos en los que se segrega la enfermedad permitirá hacer un análisis de ligamiento para determinar la posición cromosómica del locus de la enfermedad.
El investigador que realiza mapeo de genes debe analizar varios árboles genealógicos relacionados con una enfermedad particular y seleccionar un segmento genético con alelos identificables y localización cromosómica previamente definida, denominado marcador, presumiblemente vecino del gen de la afección estudiada. Posteriormente el investigador examinará la cosegregación de los alelos de la enfermedad y del marcador, y contará el número de descendientes que presentan recombinación entre los alelos del gen y del marcador. Cuantos menos recombinantes encuentre, mayores son las probabilidades de determinar la ubicación del gen, la cual se denomina locus (plural: loci). En el caso del gen de la 21-hidroxilasa, el ligamiento con el locus HLA-B facilitó la ubicación de este gen y actualmente permite realizar estudios indirectos para determinar a portadores de la mutación 17.
Una vez concluida la determinación del locus de un gen implicado en una enfermedad, se debe establecer la posición física del locus, aislar el gen en una clona manipulable (clonación) y determinar las mutaciones causantes del fenotipo patológico. Estos procedimientos, conocidos como clonamiento posicional, se describirán en la sección del Proyecto del Genoma Humano.
BASES FÍSICAS DE LA INFORMACIÓN GENÉTICA Y DOGMA CENTRAL DE LA BIOLOGÍA
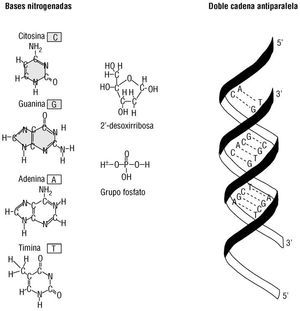
Toda la conceptualización anterior se puede ubicar en un contexto material. La base física de la información genética es el ácido desoxirribonucleico o ADN 18,19. Este polímero está constituido por desoxirribonucleósidos fosfato que pueden contener cuatro tipos de bases nitrogenadas: dos pirimidínicas denominadas citosina (C) y timina (T) y dos purínicas, guanina (G) y adenina (A) 20,21. Estos nucleótidos se enlazan en diversas secuencias de longitudes variables para constituir un gen. Antes del estudio del gen, deben analizarse algunos aspectos adicionales de este ácido. El ADN está constituido por una hélice de doble cadena. Las cadenas tienen una orientación determinada por el átomo de carbono del azúcar desoxirribosa (el que da nombre a la molécula), que participa en el enlace fosfato en el polímero. Por convención, y por otras razones bioquímicas, se determina que el extremo inicial del polímero exhibe un fosfato libre enlazado con el carbono 5' del azúcar y que éste termina en un nucleótido que exhibe un azúcar con un enlace de fosfato libre en el extremo 3' (orientación 5' a 3'). El resto de azúcares en el interior del polímero tienen enlaces fosfodiéster 3'-5' que generan el esqueleto del polímero. Las bases nitrogenadas no participan en este esqueleto. La segunda cadena se alinea con su contraparte para formar la doble hélice, en la orientación opuesta (3' a 5'), por lo que esta hélice se denomina antiparalela 20,21. Las hélices se alinean entre sí por la capacidad de las bases nitrogenadas de ambas cadenas, localizadas en el interior de la hélice, para generar puentes de hidrógeno que dan soporte a la unión. Esta interacción de bases está gobernada por el principio de complementariedad de James D. Watson y Francis H. C. Crick, elucidado por los mismos autores que describieron la estructura de esta molécula de la herencia en 1953 20,21. Este principio establece que al frente de una C habrá una G formando tres puentes de hidrógeno y que al frente de una T habrá una A formando dos puentes de hidrógeno (fig. 5).

Fig. 5. Componentes y estructura de la doble cadena antiparalela del ADN.
Durante la replicación, proceso en el cual se duplican las cadenas del ADN para las futuras células, las cadenas originales servirán de molde para la generación de cadenas complementarias, las cuales conservan la secuencia de bases materna después de finalizado el proceso 22. De esta forma opera físicamente la transmisión de la herencia en los dos procesos de división celular mencionados arriba. La replicación ocurre en la interfase e implica un proceso denominado ciclo celular (fig. 2). En general, este proceso se inicia con una fase G1 (por gap, en inglés), en la que se preparan los componentes de la replicación y se verifica el buen estado metabólico de la célula que entrará en división. En la fase S (síntesis), las cadenas hijas del ADN son generadas 23. En este proceso se requiere la participación adicional de una ADN polimerasa, un iniciador que es una hebra muy corta de ARN (5 bases) que expone un grupo fosfato unido al carbono 3' de la ribosa (éste es un gancho generalmente usado en la polimerización de ácidos nucleicos), y los desoxinucleótidos trifosfato A, C, G, y T presentes en el núcleo 24. Todos estos componentes son requeridos en el laboratorio de medicina molecular en procesos como la reacción en cadena de la polimerasa (PCR) y la secuenciación de segmentos de ADN. Después de que el material haya sido completamente replicado, la célula entra en la fase G2, en la cual toma un tiempo para determinar su condición metabólica con el objeto de preparar a la célula para una inminente división. Si todo es correcto, la célula entra en la fase M (mitosis), en la que la célula avanza a la siguiente etapa de profase. Concluida la división, las células hijas entran nuevamente en la etapa G1 en la interfase 23,25.
No obstante la información contenida en el ADN puede ser utilizada para otro propósito: la síntesis de ARN y proteínas. La generación de hebras de ARN a partir del ADN se denomina transcripción. En este momento es preciso definir en qué consiste un gen humano desde el punto de vista molecular (fig. 6). El gen estructural es la secuencia que porta toda la información para generar una cadena de ARN que se utilizará para la generación de la maquinaria ribosomal (fábrica de proteínas) o para la producción de una proteína particular 26. Las especies de ARN que pueden ser producidas son los ARN ribosomales o ARNr 28s, 18s y 5.8s (producidas por la ARN polimerasa I) que constituyen al ribosoma 27, las especies de ARN heterogéneo nuclear que darán origen al ARN mensajero o ARNm 28 (producido por la ARN polimerasa II), y las especies pequeñas de ARN de transferencia o ARNt 29, ARNs del empalmosoma (del cual se hablará más adelante) y el ARNr 5s que también forma parte del ribosoma (producidas por la ARN polimerasa III). Las ARN polimerasas sintetizan la cadena de ARN utilizando una de las cadenas del ADN como plantilla, y la introducción de cada nucleótido en la cadena generada está gobernada por las leyes de complementariedad de Watson y Crick, aunque con una notable excepción: en la hebra de ARN se introducirá una nueva base, el uracilo (U), en reemplazo de la timina. Es preferible centrar el resto de esta descripción en los genes que producen proteínas y cuyos transcritos son ARNm.

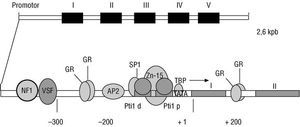
Fig. 6. Gen de la hormona del crecimiento humano. La estructura lineal representa el segmento de ADN correspondiente al gen estructural que codifica para la hormona y al segmento previo que contiene la secuencia promotora. Nótese que los genes están interrumpidos: las secuencias codificadoras o exones están representados por las cajas con números romanos, y las no codificadoras o intrones están representados por los segmentos lineales conectores. En la porción inferior se detalla una ampliación de la región promotora y se ilustra la interacción de este segmento con los factores transcripcionales representados por las estructuras ovales, las cuales permiten la expresión de este gen en la adenohipófisis. (Cortesía Biol. Aurelio Álvarez.)
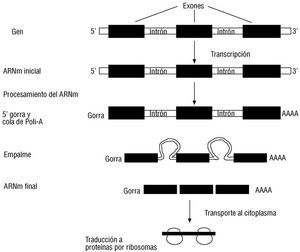
Como se ilustra en la figura 6, nuestros genes son muy particulares. Están constituidos por secuencias útiles para la producción de una proteína que se conoce como exones y por secuencias intercaladas denominadas intrones 30. La expresión armoniosa de genes en diferentes tejidos y en distintas etapas del desarrollo requiere de un complejo sistema de regulación de la expresión 31,32. De esta forma se logra que la miosina se produzca en la fibra miocárdica, mientras la queratina se produce en la piel. Para lograr en parte este control, el gen estructural posee secuencias adicionales generalmente localizadas fuera del gen estructural, denominadas elementos de regulación, que pueden ser identificadas y capturadas por proteínas denominadas factores de transcripción. Estos factores modulan la producción de una hebra particular de ARNm segúnla función del gen en el tejido específico, el estado metabólico de la célula, y según las señales recibidas en el entorno celular interno y las señales provenientes del exterior (como las hormonas). Entre los elementos de la regulación se pueden distinguir los promotores, generalmente ubicados enfrente del gen 33. Los otros elementos pueden estar en el interior del gen cerca del gen, o incluso a gran distancia del gen y pueden funcionar como aumentadores o silenciadores de la expresión génica 34. Algunas secuencias pueden activarse por la presencia de una hormona o un segundo mensajero como el AMP cíclico 35,36. Estas secuencias se denominan elementos de respuesta. Todo este sistema de botones de control, enciende y apaga genes según el tejido que se analice. También debe considerarse que el ARNm humano requiere un proceso de maduración (fig. 7) en el que los intrones son removidos por los empalmosomas (complejos de proteínas y pequeños ARNs) que ayudan a la identificación, a la remoción de los intrones y a la reunión y al ligamiento delos exones dentro del núcleo 37. Simultáneamente, los ARNm sufren dos modificaciones adicionales: la adición de una gorra ( cap en inglés) 38 en su extremo 5' y de una cola en el otro extremo 39. La gorra consiste en la adición de un nucleótido de guanina en un curioso enlace fosfodiéster 5'-5' con la primera base de la hebra y metilaciones en este residuo y en las tres primeras bases de la hebra. Esta gorra será útil para anclar a la hebra de ARNm en la maquinaria ribosomal. También a la mayoría de hebras de ARNm se les adiciona una cola de varias adeninas (poli-A) en el extremo 3', la cual ha sido muy útil para los biólogos moleculares durante la pesca de genes. Como se ve, el concepto de gen puede abarcar más que la secuencia definida como «estructural» y los daños o mutaciones en el gen o en sus elementos de empalme y regulación pueden producir enfermedades. A este concepto de gen se le puede denominar gen funcional.

Fig. 7. Maduración del ARNm. Antes que el ARN sea capturado por el aparato ribosomal para la síntesis de una proteína, esta molécula sufre un proceso de maduración en el cual se observan tres acontecimientos: se agrega una gorra en el extremo 5', se añade una cola de residuos de adenina en el extremo 3' y se remueven los intrones para permitir el empalme de los exones. Después del proceso, el ARNm abandona el núcleo y es capturado por los ribosomas.
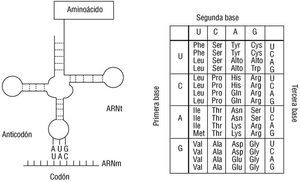
El último proceso es la producción de una proteína, una vez que la transcripción de ARN ha sido producida. Este proceso se llama traducción. Hasta ahora no se ha hecho mención alguna de los sistemas de lenguaje utilizados en la información genética, que particularmente en este punto adquieren gran relevancia. La transcripción de ARNm posee una secuencia de determinada longitud con una secuencia de bases nitrogenadas muy particular para la producción de cada proteína. Las palabras de estas hebras están formadas por tres bases consecutivas. A este triplete se le denomina codón 40. Cada codón implica la presencia de un aminoácido particular que también tiene un orden secuencial. Con la única posibilidad de generar palabras en un idioma que sólo cuenta con 4 letras, el número total de éstas es de 64 (4 3) (fig. 8). De estas 64 palabras, el idioma toma tres que significan punto final (UAG, UGA y UAA). Restan 61 palabras que deben tener correspondencia con el lenguaje de las proteínas que consta de 20 palabras (aminoácidos) 41. En este sentido, la traducción es algo parecido a buscar el equivalente de las 28 letras del castellano con los aproximadamente 50.000 ideogramas del idioma chino. Un verdadero trabajo de traducción. Sin embargo, la equivalencia de codones con aminoácidos es notable, y las equivalencias están detalladas en el código genético (fig. 8) 41. Obviamente hay palabras redundantes y algunos aminoácidos están representados por más de un codón. Por esta característica se dice que el código genético es degenerado.

Fig. 8. Estructura del ARNt y código genético. El ARNt es una molécula adaptadora que simultáneamente carga un aminoácido e identifica el codón correcto para aquel en la hebra de ARNm dentro del ribosoma. Para esta tarea, la molécula adopta una morfología de la cual destaca la burbuja en la porción inferior, donde se localiza el anticodón con una secuencia de bases complementaria al codón expuesto en el ARNm. La tabla del código genético muestra las equivalencias entre codones y aminoácidos, que se requiere para la producción precisa de las proteínas. Para identificar los codones, simplemente se localizan las tres letras del codón de forma ordenada y se determina el aminoácido correspondiente. Obsérvese que algunos aminoácidos están representados por más de un codón (p. ej,. la serina).
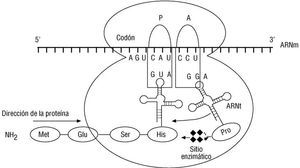
El código genético utilizado por la bacteria y los eucariontes es idéntico, pero la mitocondria, el organelo celular encargado de la provisión de energía en la célula, tiene su propio genoma, que es circular y tiene un código genético diferente. Los ARNt leerán el mensaje y facilitarán la generación de una cadena polipeptídica cuya secuencia está determinada en el gen y su correspondiente ARNm. Esta molécula es un adaptador y, para esto, posee por lo menos dos sitios funcionales importantes en su estructura bidimensional en forma de trébol (fig. 8). En su porción inferior, se presenta una secuencia particular que establecerá una unión temporal con el codón del ARNm siguiendo los principios de Watson y Crick. Este segmento de tres bases se denomina anticodón 42. En su extremo 3', esta molécula portará un aminoácido que será integrado en la proteína. Unas enzimas denominadas aminoacil-ARNt transferasas se encargan de determinar el anticodón del ARNt y cargarlo con el aminoácido adecuado, según los principios del código genético. Establecidas las reglas del código genético, se puede analizar la maquinaria traduccional que genera las proteínas (fig. 9).

Fig. 9. Traducción. La hebra de ARNm ha sido capturada por el ribosoma y en su interior se observan dos sitios: el sitio P (peptidil), ocupado por un ARNt que carga a la proteína en crecimiento, y el sitio A (aminoacil), al cual ingresa un ARNt cargado con un aminoácido particular correspondiente al codón expuesto en éste mismo sitio. Para concluir la adición del nuevo aminoácido a la proteína, el ribosoma cuenta con una actividad enzimática en la zona intermedia de los sitios A y P, en la que se realiza la unión peptídica entre los aminoácidos cargados por las dos moléculas de ARNt.
La síntesis de estas moléculas se realiza en tres pasos. Durante la iniciación, la hebra de ARNm madura abandona el núcleo y es capturada por los ribosomas. Este proceso complejo requiere la participación de proteínas o factores adicionales que, en esencia, permiten el ensamblaje de la hebra en la subunidad menor del ribosoma (30s), de la integración del primer ARNt cargando un residuo de metionina y el acoplamiento de la subunidad ribosómica mayor (80s). El ARNt mencionado ocupa un espacio denominado P (por sitio peptidil), en el cual se engancha el péptido en crecimiento. En la vecindad inmediata, queda expuesto el segundo codón del ARNm. Esta posición se llama A (por sitio aminoacil) y será ocupada por el ARNt correspondiente al codón de esta posición. La subunidad mayor acerca los aminoácidos y establece un enlace peptídico entre el grupo carboxilo de la metionina y el grupo amino del segundo aminoácido en una reacción que requiere el consumo de la energía aportada por ATP. El dipéptido formado queda unido al segundo ARNt y se encuentra ocupando el sitio A, de tal manera que durante un proceso denominado translocación este ARNt es desplazado al sitio P, se libera el ARNt de la metionina y queda libre el espacio para el ingreso de un nuevo aminoácido, determinado esta vez por el tercer codón que queda expuesto en el sitio A. Estos procesos son facilitados por los factores de elongación y la secuencia de acontecimientos continúa hasta que la hebra de ARNm ha sido completamente traducida. En este momento un factor de liberación permite el desensamblaje de la maquinaria ribosomal y la liberación del péptido formado y del último ARNt involucrado en el proceso 43. La cadena libre será modificada por proteínas moldeantes como las chaperonas 44 o mediante la adición de grupos químicos como el metilo, acetilo, oligosacáridos, etc. Estas modificaciones ocurren principalmente en el retículo endoplásmico y en el aparato de Golgi, y dependerán tanto de la función como del destino final de la proteína recién sintetizada 45,46.
Antes de continuar, es importante hacer unas consideraciones adicionales sobre la mitocondria. Este organelo intracelular tiene su propia maquinaria genética y es capaz de replicarse de forma independiente dentro de la célula. Su material genético está constituido por una cadena circular de aproximadamente 16.500 pares de bases y contiene 37 genes, 13 de los cuales codifican para proteínas implicadas en la fosforilación oxidativa y en la producción de energía, la principal función de este organelo, en la cual participan 69 proteínas 47. Los restantes 24 genes codifican para dos ARNr y 22 ARNt. Con estos genes y productos proporcionados por los genes del núcleo, la mitocondria puede realizar su propia replicación, transcripción y traducción. Durante la generación de un nuevo individuo en la fecundación, las mitocondrias son principalmente proporcionadas por el óvulo, ya que las mitocondrias del espermatozoide están ubicadas en el cuello, una estructura que contribuye con un número muy escaso de estos organelos en el nuevo ser 46. Por esta razón, la herencia mitocondrial también se conoce como herencia materna 48. Debido a la capacidad de estos organelos de replicarse y segregarse a las células hijas de forma independientemente, la proporción de mitocondrias portadoras de una mutación patológica generalmente varía dentro de los diversos tejidos del afectado. Esta condición, denominada heteroplasmia, tiene un papel importante en la variabilidad y severidad de la enfermedad mitocondrial.
EL PROYECTO DEL GENOMA HUMANO
La primera descripción de la participación de los genes en la herencia y en la enfermedad humana la realizó Archibald Garrod en colaboración con el biólogo William Bateson en 1902. Este doctor inglés estudió a una familia con alcaptonuria [MIM 203500], un trastorno del metabolismo del aminoácido fenilalanina caracterizado por oscurecimiento de la orina, artritis, ocronosis y enfermedad prematura de las arterias coronarias 49. Ambos llegaron a la conclusión de que el trastorno se transmitía de forma autosómica recesiva y acuñaron el término «errores innatos del metabolismo», el cual condiciona el comportamiento anormal del sustrato y del producto cuando existe un bloqueo enzimático en una ruta metabólica, determinado por la presencia de un gen defectuoso para la síntesis adecuada de una enzima 50. En ese mismo año, Walter S. Sutton y Theodor Bovery lanzaron una teoría que implicaba a los cromosomas como vehículos transmisores de la herencia 4, hipótesis que sería verificada por el descubrimiento de Watson y Crick 50 años después. Hasta ese momento la naturaleza de la herencia humana no había despertado gran interés en los investigadores médicos, y tal vez el descubrimiento más importante es el realizado em 1949 por Linus Pauling, quien describe que la anemia de células falciformes es una «enfermedad molecular» debida a una alteración de la cadena b-globina y que resulta en una hemoglobina inestable 51. Después del descubrimiento y la descripción del ADN como la molécula de la herencia y portadora de los genes por Watson y Crick a mediados del siglo xx, los nuevos descubrimientos en el campo de la genética médica se realizaron en forma pausada. Tres años después del descubrimiento de Watson y Crick, Hin Tjio y Levan determinaron el número de cromosomas en la especie humana 52, y en 1959 Lejeune et al determinaron que el síndrome de Down estaba causado por una trisomía 53. A partir de este momento la genética clínica se consolida como una disciplina médica. Sin embargo, la mayoría de los nuevos descubrimientos siguen surgiendo del área de la biología y no son rápidamente incorporados en la nueva especialidad médica. Durante los años sesenta y setenta, los trabajos de Jacob y Manod sobre la regulación de la expresión génica en la bacteria 54, el descubrimiento de las enzimas de restricción 55,56, la aparición de los métodos de clonación de segmentos de ADN en plásmidos bacterianos 57, y la descripción de los métodos de secuenciación del ADN realizada independientemente por Maxam y Gilbert 58, y por Sanger et al 59 en 1977 aceleraron la comprensión de la estructura de los genes y de la fisiología genética. El análisis de hibridación de cadenas sencillas de ADN descrito por Southern en 1975 acercó nuevamente la biología molecular y la genética médica 60. En 1978 Kan y Dozi describieron polimorfismos genéticos ligados con la mutación determinante de la anemia de células falciformes mediante análisis de tipo Southern, la cual es útil para el diagnóstico prenatal de la enfermedad 61.
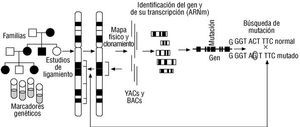
Al inicio de los años ochenta, ya existía un panorama favorable para imaginar que la secuencia completa del genoma humano podía ser determinada y que la ubicación de todos los genes en los cromosomas permitiría realizar una cartografía completa del genoma humano. Sin embargo, todavía faltaba una invención que potenciaría enormemente la capacidad de analizar secuencias de ADN: la reacción en cadena de la polimerasa (PCR) 62. Esta admirable técnica consiste en la amplificación geométrica en un microtubo, de una secuencia seleccionada del genoma mediante una síntesis artificial de pequeños segmentos de ADN (entre 100 y 2.000 pares de bases), usando la enzima y los componentes que la célula requiere para este proceso. A finales de la década de los ochenta, el Departamento de Energía (DOE) y los Institutos Nacionales de Salud (NIH) de Estados Unidos unieron sus recursos para lanzar el Proyecto del Genoma Humano en otoño de 1990, liderado inicialmente por Watson, y en el cual se esperaba la participación de todos los laboratorios existentes de genética molecular humana 63. La misión del proyecto era precisamente determinar la secuencia de todo el genoma humano y facilitar la ubicación de los aproximadamente 70.000 genes de nuestra especie. Los avances fueron muy rápidos. En los primeros años del proyecto, centros como el grupo francés CEPH (Centre d'Études du Polymorphisme Humain) 64,65 y las Universidades de Utah y de Washington en San Luis (Missouri), de Estados Unidos, etc. crearon librerías del genoma humano disponibles en diferentes tipos de vectores como cósmidos, cromosomas artificiales de levadura (YAC), cromosomas artificiales de bacteria (BAC), etc. Una librería genómica contiene todos los fragmentos resultantes del fraccionamiento del genoma humano con enzimas de restricción, clonados en los vectores mencionados 66, pero en esta librería todo el genoma está disperso, como las piezas de un rompecabezas esparcidas sobre una mesa. Los laboratorios participantes en el esfuerzo se dedicaron a identificar clonas y generar cóntigos con los fragmentos de interés mediante una serie de metodologías denominadas clonamiento posicional 67 (fig. 10). Un cóntigo es una secuencia resultante del sobrelapamiento de fragmentos clonados en los cuales puede residir el gen. Las secuencias de ADN utilizadas para seleccionar las clonas han sido determinadas previamente por el investigador que realizó los estudios de ligamiento en las familias con un padecimiento mendeliano, quien identifica los segmentos de ADN de secuencia y ubicación cromosómica conocida más apropiados para pescar estas clonas mediante hibridación o amplificación por PCR 67. Los cóntigos generados son utilizados para generar mapas físicos. Un mapa físico es la representación ordenada de los segmentos clonados que cubren una porción de un cromosoma y en el cual se determina la localización exacta en un gen. Este mapa tiene estrecha correlación con el mapa genético, determinado por los estudios de ligamiento como ya se ha mencionado.

Fig. 10. Clonamiento posicional. Esta estrategia para el aislamiento de un gen se inicia con la colección de varios árboles genealógicos de sujetos con una enfermedad o rasgo particular, para la realización de estudios de ligamiento con marcadores genéticos polimórficos. Con los datos obtenidos, se determina una ubicación cromosómica, que es analizada de forma más detallada mediante la construcción de un mapa físico de la zona sospechosa. Este mapa se realiza con fragmentos de DNA conservados en librerías genómicas clonadas en YACs, BACs, etc. Los mismos marcadores utilizados en el estudio de ligamiento permiten identificar las clonas de interés, y generalmente se obtienen múltiples fragmentos equivalentes a las piezas de un rompecabezas. Algunos análisis adicionales permiten determinar los extremos de solapamiento entre los diferentes fragmentos para construir un segmento más largo de información genética continua que se denomina cóntigo (incluso se pueden construir cóntigos grandes con cóntigos predeterminados). Una vez realizados los cóntigos, se hace un análisis de su información genética para determinar posibles regiones involucradas en la producción de ARNm (secuencias transcriptoras) que corresponden a genes potenciales. El estudio de estas secuencias permitiría ubicar a un gen fisiológicamente relacionado con el trastorno genético que originó el estudio. En este momento se debe comprobar de forma amplia que el segmento está involucrado en la transcripción (incluso se comprueba que el mismo segmento se expresa en otras especies de animales). Por último se buscan mutaciones a lo largo de la secuencia genética en los sujetos afectados para concluir simultáneamente cuál es el gen responsable del fenotipo y el tipo de daño genético responsable de éste. (Modificada de Gelehrter TD, Collins FS, Ginsburg D. Principles of medical genetics [2.a ed.]. Baltimore: Williams and Wilkins, 1998; 224.)
Los expertos en informática, incluidos en este proyecto desde su inicio, se han encargado de anotar los descubrimientos y alimentar diversos bancos de datos disponibles a toda la comunidad científica. Así, el Gene Data Bank ( www.ncbi.nlm.nih.gov/genbank) y otras fuentes ofrecen información sobre las secuencias proporcionadas de forma altruista por cualquier investigador del Proyecto del Genoma Humano. El avance en la cobertura del mapa físico de todo el genoma humano y la velocidad de descubrimiento de genes eran impresionantes a finales de los noventa, y los expertos preveían que las metas de la tarea podrían alcanzarse más rápidamente de lo planeado 68. Al campo de trabajo se unieron expertos en procesos de robótica y microtecnología que produjeron artefactos para la manipulación masiva de muestras y la secuenciación en procesos industriales de alto rendimiento (high-throughput technologies). Como resultado, en marzo del presente año J. Craig Venter, fundador de la compañía Celera e investigador inicialmente asociado al Proyecto del Genoma Humano, anuncia que el 99% del genoma humano ha sido secuenciado en los dos componentes de la cadena de ADN en su industria privada. La metodología utilizada por el grupo Celera para realizar la secuenciación del genoma humano, denominada secuenciación por escopetazo, fue inicialmente probada para secuenciar el genoma de la mosca de la fruta 69,70. Esta metodología consiste en tomar el genoma de un individuo y fragmentarlo en trozos pequeños de 100.000 a 200.000 pb) que se clonan en el vector BAC y que fácilmente se reproducen en bacteria. Se diseñan iniciadores para la secuenciación del inserto humano del BAC con las secuencias del vector cercanas a los dos extremos de clonación, y un proceso de secuenciación automático de muy alto rendimiento determina la secuencia del fragmento clonado. Esta información va siendo recolectada por un ordenador en el momento de ser generada. El ordenador está alimentado con un programa que ordena todas las nuevas secuencias generadas y produce un alineamiento correcto de los insertos relacionados para la generación de cóntigos en una forma acelerada. A fin de corroborar un perfecto ordenamiento, el sistema determina la secuencia de las dos cadenas antiparalelas del ADN, de tal manera que hay un criterio adicional para confirmar la exactitud del cóntigo. Esto es particularmente importante para el ensamblaje de regiones que presentan secuencias repetitivas, las cuales siempre han representado un obstáculo para el ordenamiento de las secuencias de un cóntigo. Sólo restan dos tareas: completar el ordenamiento de todos los segmentos secuenciados en los 23 cromosomas y asignar la ubicación de los genes que aún no han sido localizados. Las estadísticas consignadas en el Catálogo de la Herencia Mendeliana del Hombre revelan que para junio del presente año se han establecido los loci de 8.424 genes (tabla 1). El 26 de junio de este año, se logra un acuerdo entre los consorcios públicos y privados participantes en este proyecto, liderados por el Proyecto del Genoma Humano del DOE y el NIH, el grupo Celera y la fundación Wellcome Trust de Inglaterra, para dar a conocer de forma conjunta los resultados de las secuencias humanas secuenciadas de manera precisa y ensambladas, las cuales serán vertidas a una base de datos pública. Con estos avances y con los acuerdos logrados, es predecible que en uno o dos años más el mapa del genoma humano esté completamente determinado y, tal vez, disponible al público en los ordenadores.

MEDICINA MOLECULAR
La medicina molecular es una nueva concepción en la cual la salud y la enfermedad están determinadas por un universo cambiante y de arquitectura flexible, constituido por moléculas como ácidos nucleicos, proteínas, hidratos de carbono, lípidos, agua, sales, metales, etc., que generan subsistemas termodinámicos de complejidad creciente conocidos como célula, tejido, órgano, sistema y cuerpo. Con esta concepción, se intenta entender la homeostasis celular, tisular y sistémica, así como definir fenómenos como el origen, el crecimiento, la reproducción y la muerte al nivel más microscópico que la tecnología actual permite. Este concepto también intenta explicar cómo algunos mecanismos internos o externos producen la enfermedad por ausencia de algunos de sus componentes, como en los errores innatos del metabolismo o en las deficiencias hormonales; por alteraciones en las frecuencias o momentos en los que estos mecanismos ocurren en el interior del organismo, como en el cáncer, o por la aparición de episodios nuevos y estocásticos que desde el exterior modifican la homeostasis molecular, como las carencias nutricionales y los agentes infecciosos o tóxicos.
Es difícil que cualquiera de los acontecimientos que afectan nuestra salud escape de este marco de moléculas en movimiento, aun los difíciles procesos psicológicos. Esta concepción es muy antigua y previa a los trabajos de Garrod, pero en años recientes el Proyecto del Genoma Humano y disciplinas como la genética clínica, la bioquímica, la biología celular y otras ramas muy recientes como la genómica y la bioinformática han enriquecido de forma fabulosamente acelerada nuestro conocimiento sobre todos los eventos que ocurren en el cuerpo humano y han dado consistencia al concepto de medicina molecular.
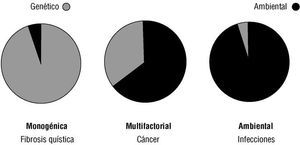
Como se ha mencionado en el párrafo anterior, las enfermedades pueden ubicarse en diferentes posiciones de un espectro determinado por las proporciones en que interactúan los factores genéticos y los ambientales (fig. 11). En un extremo estarán las dolencias debidas casi exclusivamente a un factor genético, como las enfermedades de herencia mendeliana (p. ej., la fibrosis quística), y en el otro se encontrarán aquellas afecciones generadas por el medio ambiente, como las enfermedades infecciosas. Es muy difícil encontrar ejemplos de padecimientos en los que uno de los dos componentes mencionados no desempeñe un papel mínimo para el desarrollo de la enfermedad (quizás se escapen los debidos a la violencia, sin precisar hasta qué punto existen determinantes genéticos en el comportamiento del agresor). Desde el punto de vista genético, se pueden distinguir los siguientes tipos de enfermedades (tabla 2):

Fig. 11. Componentes ambiental y genético de la enfermedad. Las enfermedades en general tienen un componente o determinante genético y otro ambiental. En un extremo del espectro, la expresión de las enfermedades mendelianas con un determinante genético principal puede ser modificada por el medio ambiente. En el otro extremo, las enfermedades ambientales, como las infectocontagiosas, tienen un componente genético que determina la susceptibilidad y resistencia al agente nocivo. La mayoría de las enfermedades crónicas (diabetes mellitus, hipertensión, cáncer, etc.) se ubican en el panorama intermedio.

Enfermedades monogénicas o mendelianas
Son aquellos padecimientos hereditarios producidos por alteración de un solo gen cromosómico. Actualmente hay más de 700 rasgos mendelianos descri-tos en el Catálogo de la Herencia Mendeliana del Hombre 5, de los cuales una gran proporción pueden tener repercusiones patológicas (algunos rasgos, como los grupos sanguíneos, no tienen significación patológica). Afecciones que comprometen el sistema cardiovascular, como el síndrome de Marfan [MIM 154700], los síndromes de Ehlers-Danlos I [MIM 130000], III [MIM 130020] y IV [MIM 130050], el síndrome de Noonan [MIM 163950], el síndrome de Holt-Oram [MIM 142900], los síndromes de arritmia como el síndrome del Q-T largo [MIM 192500] y el síndrome de Brugada 71 [MIM 601144] y los cuadros de hipercolesterolemia familiar [MIM 143890], pertenecen a este grupo. Estos cuadros se dividen en cuatro grupos según el tipo de trasmisión del defecto hereditario, el cual está determinado por la localización del gen y por la naturaleza del producto generado. Si el gen mutado está localizado en un cromosoma no sexual (autosoma), en el cromosoma X o en el cromosoma Y, producirá, respectivamente, una enfermedad autosómica, ligada al cromosoma X o ligada al cromosoma Y. Según la naturaleza del producto generado, las enfermedades podrán ser dominantes o recesivas. En las primeras, la herencia de una sola copia mutada de los dos alelos paternos es suficiente para que la enfermedad se manifieste, como ocurre en el síndrome de Marfan. Aunque no es posible generalizar, este tipo de trasmisión hereditaria se observa en las enfermedades relacionadas con proteínas estructurales, como es el caso de la fibrilina 1 en el síndrome mencionado. En los cuadros recesivos, se requiere la herencia de los dos alelos paternos en sus versiones mutadas para que ocurra la enfermedad, como en la enfermedad de Pompe (enfermedad de almacenamiento del glucógeno tipo II) [MIM 232300], una deficiencia de la enzima lisosomal a-1,4-glucosidasa ácida, cuyo gen está localizado en el brazo largo del cromosoma 17. En esta enfermedad se produce una acumulación de glucógeno que es la causa de signos como macroglosia, hepatomegalia y miocardiopatía.
Los trastornos ligados al cromosoma X también pueden presentar patrones dominantes o recesivos, siendo los últimos los más frecuentes. Entre los síndromes recesivos ligados al X, se encuentran tres enfermedades producidas por mutaciones en el gen de la distrofina: las distrofias musculares de Duchenne y de Becker y la miocardiopatía ligada al cromosoma X [MIM 310200]. La distrofina es una proteína que sirve como puente de anclaje entre la actina y la membrana plasmática o sarcolema. En este tipo de enfermedades recesivas, el padecimiento hereditario afecta a los varones que reciben el cromosoma X mutado, el cual es transmitido por una portadora que no sufre el cuadro (aunque muchas veces presenta alteraciones fisiológicas, como la elevación en plasma de enzimas musculares como la creatinfosfocinasa). Entre las enfermedades dominantes ligadas al cromosoma X, la más frecuente es el raquitismo resistente a la vitamina D [MIM 307800]. Estas enfermedades afectan a la portadora y a sus hijos de ambos sexos que reciben el gen mutado, pero la severidad del cuadro es más leve en las portadoras. Finalmente, las enfermedades ligadas al cromosoma Y, aún no descritas en la especie humana, serían de carácter dominante y presentarían un patrón de transmisión de varón a varón. Éstas afectarían a genes responsables de la espermatogénesis. Aunque en un pasado no muy distante era imposible la transmisión genética de estas alteraciones, debido al cuadro de infertilidad del afectado, en años recientes el uso del método de reproducción asistida denominado inyección intracitoplásmica de esperma (ICSI) 72, en el que un espermatozoide obtenido del testículo del varón infértil es introducido en un óvulo, permitiría la procreación de varones con el defecto hereditario (que probablemente sería la única manifestación de la enfermedad).
Cromosomopatías
Estas enfermedades, como se mencionó al principio, se deben a una alteración del número de cromosomas (aneuploidía) o de la estructura de éstos. Son muy pocos los trastornos de este tipo que son compatibles con la vida posnatal. Entre las aneuploidías, las más frecuentes son las trisomías 21 (síndrome de Down) 73, 18 (síndrome de Edwards) 74 y 13 (síndrome de Patau) 75. Las tres cursan con defectos cardíacos que limitan la supervivencia. En el síndrome de Turner, una monosomía del cromosoma X (45, X0), las anormalidades cardíacas están presentes entre un 25 y un 50% de los pacientes, observándose frecuentemente coartación de la aorta 76. Entre las alteraciones de la estructura cromosómica más frecuentes se pueden mencionar las deleciones del brazo corto del cromosoma 4 o 4p- (síndrome de Wolf-Hirschhorn) 77 y la del brazo corto del cromosoma 5 o 5p- (síndrome cri du chat) 78. En ambos casos los defectos del tabique interventricular, la persistencia del ducto arterioso y la estenosis pulmonar son frecuentes.
Enfermedades mitocondriales
Las mutaciones de los genes mitocondriales pueden producir alteraciones del metabolismo aeróbico y muchos de estos cuadros afectan al corazón, como el síndrome de Kearns-Sayre [MIM 530000], caracterizado por miopatía generalizada, oftalmoplejía, retinopatía, trastornos de la conducción cardíaca y miocardiopatía dilatada 79, y el síndrome MELAS (miopatía mitocondrial, encefalopatía, acidosis láctica y paraplejía) [MIM 540000], en el que se han descrito casos de miocardiopatía hipertrófica y dilatada, y la miocardiopatía idiopática dilatada mitocondrial [MIM 510000] debida a mutaciones en el gen tARN-isoleucina, etc.
Enfermedades poligénicas o multifactoriales
Hay muchos tipos de caracteres hereditarios cuyo mecanismo de transmisión no se puede encajar en las leyes mendelianas, tales como la inteligencia, la estatura, el comportamiento, etc. Los mecanismos que determinan la herencia de estos caracteres están determinados por la acción aditiva de muchos genes y, además, la expresión está influida por el medio ambiente. Muchos trastornos crónicos del adulto, como la hipertensión arterial esencial 80, la diabetes mellitus no insulino-dependiente 81 y algunas malformaciones congénitas, particularmente las cardiopatías congénitas aisladas 82, presentan este tipo de herencia. Para estas últimas se ha descrito que se deben a fallos en seis pasos críticos del desarrollo embrionario: migración anormal del tejido ectomesenquimal, flujo sanguíneo intracardíaco anormal, crecimiento anormal focalizado, anormalidad de la matriz extracelular, defectos de los cojinetes endocárdicos y defectos de la formación del tubo cardíaco. Todos estos defectos pueden producir desde cuadros relativamente leves, como la persistencia del conducto arterioso, hasta severas patologías como la tetralogía de Fallot o la transposición de grandes vasos 83.
En este tipo de enfermedades se observa frecuentemente la agrupación del defecto en una familia y la predilección del defecto por un sexo, como es el caso del sexo femenino para este tipo de alteraciones cardíacas. El modelo matemático de Fisher para determinar la aparición de estos defectos establece que el efecto aditivo de la acción poligénica debe cruzar un umbral para precipitar la aparición del cuadro. Cada enfermedad de este tipo requiere de un análisis especial para determinar el riesgo de recurrencia, y el tema de las malformaciones cardíacas congénitas ha sido objeto de revisión por varios autores entre los que destacan Edwards 83 y Nora 84 et al.
Enfermedades genéticas de células somáticas (alteraciones genéticas acumulativas)
El prototipo claro de este tipo de trastornos es el cáncer. En esta enfermedad, la célula maligna resulta de la acumulación de diferentes mutaciones en su genoma a lo largo del tiempo 85. En la mayoría de los casos estas mutaciones comienzan a acumularse después de la fecundación y están restringidas a las clonas celulares de la célula afectada, por lo cual tiene un carácter somático. Por ejemplo, en la leucemia mieloide crónica, uno de los acontecimientos iniciales debe ser la adquisición del cromosoma Filadelfia (una translocación balanceada entre los cromosomas 9 y 22 que resulta en un gen híbrido de características oncogénicas: el gen bcr-abl). Este rearreglo sólo tiene repercusiones en la línea progenitora mieloide, y sobre ésta se acumulan nuevas mutaciones que finalmente determinan el fenotipo maligno 86. Sin embargo, en raras ocasiones la primera mutación con efecto carcinogénico puede haber sido transmitida por uno de los gametos y, además de poderse transmitir a la próxima generación, está presente en todas las células del cuerpo, como ocurre en los casos hereditarios de retinoblastoma o del tumor de Wilms 87. AGRADECIMIENTOS
El Dr. A. Rojas recibe apoyo del programa Cátedras Patrimoniales II del Consejo Nacional de Ciencia y Tecnología de México (CONACYT) y la Dra. R. Ortiz López está patrocinada por el programa de Repatriación de Científicos Mexicanos del CONACYT.
Bibliografía
[1]
Human genetics: problems and approaches (3.
[2]
Moorhead PS, Nowell PC, Mellman WJ, Battips DM, Hungerford DA..
Chromosome preparations of leukocytes cultured from human peripheral blood..
Exp Cell Res, (1960), 20 pp. 613-616
[3]
Congenital heart disease. En: Rimoin DL, Connor JM, Pyeritz RE, editores. Emery and Rimoin's principles and practice of medical genetics (3.
[4]
Classic papers in genetics. Englewood Cliffs: Prentice-Hall, 1959.
[5]
McKusick-Nathans Institute for Genetic Medicine, Johns Hopkins University (Baltimore, MD) and National Center for Biotechnology Information, National Library of Medicine (Bethesda, MD), 2000. World Wide Web URL: http: //www.ncbi.nlm.nih.gov/omim/.
[6]
Mulley JC, Haan EA, Sheffield LJ, Sutherland GR..
Recombination frequencies between Duchenne muscular dystrophy and intragenic markers in multigeneration families..
Hum Genet, (1988), 78 pp. 296-297
[7]
Towbin JA, Ortiz-López R..
X-linked dilated cardiomyopathy..
N Engl J Med, (1994), 330 pp. 369-370
[8]
Pyeritz RE..
The Marfan syndrome..
Ann Rev Med, (2000), 51 pp. 481-510
[9]
Ackroyd RS, Finnegan JA, Green SH..
Friedreich's ataxia. A clinical review with neurophysiological and echocardiographic findings..
Arch Dis Child, (1984), 59 pp. 217-221
[10]
Pérez Álvarez F..
Arrhythmia-hyperkalemia in a newborn infant with congenital adrenogenital syndrome. Usefulness of cardioversion..
An Esp Pediatr, (1986), 25 pp. 359-362
[11]
Dupont B, Oberfield SE, Smithwick EM, Lee TD, Levine LS..
Close genetic linkage between HLA and congenital adrenal hyperplasia (21-hydroxylase deficiency)..
Lancet, (1977), 2 pp. 1309-1312
[12]
Morgan TH..
Sex-limited inheritance in Drosophila..
Science, (1910), 32 pp. 120-122
[13]
The mechanism of Mendelian heredity. Nueva York: Henry Holt & Co., 1925.
[14]
Donis-Keller H, Green P, Helms C, Cartinhour S, Weiffenbach B, Stephens K..
A genetic linkage map of the human genome..
Cell, (1987), 51 pp. 319-337
[15]
Dib C, Faure S, Fizames C, Samson D, Drouot N, Vignal A et al..
A comprehensive genetic map of the human genome based on 5,264 microsatellites..
Nature, (1996), 380 pp. 152-154
[16]
How do you compute a lod score? Nat Genet 1995; 11: 354-355.
[17]
Klouda PT, Harris R, Price DA..
Linkage and association between HLA and 21-hydroxylase deficiency..
J Med Genet, (1980), 17 pp. 337-341
[19]
Avery OT, MacLeod CM, McCarty M..
Studies on the chemical nature of the substance inducing transformation of pneumococcal types. Induction of transformation by a deoxyribonucleic acid fraction isolated from pneumococcus type III..
J Exp Med, (1944), 79 pp. 137-158
[20]
Watson JD, Crick FH..
Genetical implications of the structure of deoxyribonucleic acid..
Nature, (1953), 171 pp. 964-967
[21]
Watson JD, Crick FH..
Molecular structure of nucleic acids: a structure for deoxyribose nucleic acid..
Nature, (1953), 171 pp. 737-738
[22]
Meselson M, Stahl FW..
The replication of DNA in E. coli..
Proc Nat Acad Sci USA, (1958), 44 pp. 671-682
[24]
Kornberg A..
Biologic synthesis of deoxyribonucleic acid. 1959 Nobel prize lecture..
Science, (1960), 131 pp. 1503-1508
[25]
Hartwell LH, Culotti J, Pringle JR, Reid BJ..
Genetic control of the cell division cycle in yeast..
Science, (1974), 183 pp. 46-51
[27]
Noller HF..
Structure of ribosomal RNA..
Annu Rev Biochem, (1984), 53 pp. 119-162
[28]
Hoagland MB, Stephenson ML, Scott JF, Hecht LI, Zamecnik PC..
A soluble ribonucleic acid intermediate in protein synthesis..
J Biol Chem, (1958), 231 pp. 241-257
[29]
Kim SH, Suddath FL, Quigley GJ, McPherson A, Sussman JL, Wang AH..
Three-dimensional tertiary structure of yeast phenylalanine transfer RNA..
Science, (1974), 185 pp. 435-440
[30]
Breathnach R, Chambon P..
Organization and expression of eucaryotic split genes coding for proteins..
Annu Rev Biochem, (1981), 50 pp. 349-383
[31]
Holliday R, Pugh JE..
DNA modification mechanisms and gene activity during development..
Science, (1975), 187 pp. 226-232
[32]
Youderian P, Bouvier S, Susskind MM..
Sequence determinants of promoter activity..
Cell, (1982), 30 pp. 843-853
[32]
Moldave K..
Eukaryotic protein synthesis..
Annu Rev Biochem, (1985), 54 pp. 1109-1149
[34]
Muller MM, Gerster T, Schaffner W..
Enhancer sequences and the regulation of gene transcription..
Eur J Biochem, (1988), 176 pp. 485-495
[35]
Epstein W, Rothman-Denes LB, Hesse J..
Adenosine 3':5'-cyclic monophosphate as mediator of catabolite repression in Escherichia coli..
Proc Natl Acad Sci U S A, (1975), 72 pp. 2300-2304
[36]
Simpson RB..
Interaction of the cAMP receptor protein with the lac promoter..
Nucleic Acids Res, (1980), 8 pp. 759-766
[39]
Lim L, Canellakis ES..
Adenine-rich polymer associated with rabbit reticulocyte messenger RNA..
Nature, (1970), 227 pp. 710-712
[40]
Nirenberg MW, Mattchaei JH..
The dependence of cell-free protein synthesis in E. coli upon naturally occurring or synthetic polyribonucleotides..
Proc Natl Acad Sci Wash, (1961), 47 pp. 1588
[41]
Crick FH, Barnett L, Brenner S, Watts-Tobin RJ..
General nature of the genetic code for proteins..
Nature, (1961), 192 pp. 1227-1232
[42]
Crick FH..
Codon-anticodon pairing: the wobble hypothesis..
J Mol Biol, (1966), 19 pp. 548-455
[43]
Merrick WC..
Mechanism and regulation of eukaryotic protein synthesis..
Microbiol Rev, (1992), 56 pp. 291-315
[44]
Mayhew M, Da Silva AC, Martin J, Erdjument-Bromage H, Tempst P, Hartl FU..
Protein folding in the central cavity of the GroEL-GroES chaperonin complex..
Nature, (1996), 379 pp. 420-426
[45]
Rapoport TA, Jungnickel B, Kutay U..
Protein transport across the eukaryotic endoplasmic reticulum and bacterial inner membranes..
Annu Rev Biochem, (1996), 65 pp. 271-303
[46]
Schatz G, Dobberstein B..
Common principles of protein translocation across membranes..
Science, (1996), 271 pp. 1519-1526
[47]
Grivell LA..
Mitochondrial DNA. Small, beautiful and essential..
Nature, (1989), 341 pp. 569-571
[48]
Ankel-Simons F, Cummins JM..
Misconceptions about mitochondria and mammalian fertilization: implications for theories on human evolution..
Proc Natl Acad Sci U S A, (1996), 93 pp. 13859-13863
[49]
Garrod AE..
The incidence of alkaptonuria: a study in chemical individuality. 1902..
Mol Med, (1996), 2 pp. 274-282
[51]
Pauling L, Itano HA, Singer SJ, Wells IC..
Sickle cell anemia: a molecular disease..
Science, (1949), 110 pp. 543-548
[52]
Hin Tjio J, Albert Levan A..
The chromosome number in man..
Hereditas, (1956), 42 pp. 1-6
[53]
Lejeune J, Gautie.r, M, Turpin R..
Etude des chromosomes somatiques de neuf enfants mongoliens..
C R Acad Sci, (1959), 248 pp. 1721-1722
[54]
Jacob F, Manod J..
Genetic regulatory mechanisms in the synthesis of proteins..
J Mol Biol, (1961), 3 pp. 318-356
[55]
Meselson M, Yuan R..
DNA restriction enzyme from E. coli..
Nature, (1968), 217 pp. 1110-1114
[57]
Rougeon F, Kourilsky P, Mach B..
Insertion of a rabbit beta-globin gene sequence into an E. coli plasmid..
Nucleic Acids Res, (1975), 2 pp. 2365-2378
[58]
Maxam AM, Gilbert W..
A new method for sequencing DNA..
Proc Natl Acad Sci U S A, (1977), 74 pp. 560-564
[59]
Sanger F, Nicklen S, Coulson AR..
DNA sequencing with chain-terminating inhibitors..
Proc Natl Acad Sci U S A, (1977), 74 pp. 5463-5467
[60]
Southern EM..
Detection of specific sequences among DNA fragments separated by gel electrophoresis..
J Mol Biol, (1975), 98 pp. 503-517
[61]
Kan YW, Dozy AM..
Polymorphism of DNA sequence adjacent to human beta-globin structural gene: relationship to sickle mutation..
Proc Natl Acad Sci U S A, (1978), 75 pp. 5631-5635
[62]
Saiki RK, Scharf S, Faloona F, Mullis KB, Horn GT, Erlich HA et al..
Enzymatic amplification of beta-globin genomic sequences and restriction site analysis for diagnosis of sickle cell anemia..
Science, (1985), 230 pp. 1350-1354
[63]
Watson ID..
The human genome project: past, present, and future..
Science, (1990), 248 pp. 44-49
[64]
NIH/CEPH Collaborative Mapping Group..
A comprehensive genetic linkage map of the human genome..
Science, (1992), 258 pp. 67-86
[65]
Dausset J, Ougen P, Abderrahim H, Billault A, Sambucy JL, Cohen D et al..
The CEPH YAC library..
Behring Inst Mitt, (1992), 91 pp. 13-20
[66]
Collins FS..
Positional cloning: let's not call it reverse anymore..
Nat Genet, (1992), 1 pp. 3-6
[67]
Gyapay G, Morissette J, Vignal A, Dib C, Fizames C, Millasseau P et al..
The 1993-94 Genethon human genetic linkage map..
Nat Genet, (1994), 7 pp. 246-339
[68]
Collins FS, Patrinos A, Jordan E, Chakravarti A, Gesteland R, Walters L..
New goals for the U.S. Human Genome Project: 1998-2003..
Science, (1998), 282 pp. 682-689
[69]
Adams MD, Celniker SE, Holt RA, Evans CA, Gocayne JD, Amanatides PG et al..
The genome sequence of Drosophila melanogaster..
Science, (2000), 287 pp. 2185-2195
[70]
Myers EW, Sutton GG, Delcher AL, Dew IM, Fasulo DP, Flanigan MJ et al..
A whole-genome assembly of Drosophila..
Science, (2000), 287 pp. 2196-2204
[71]
Chen Q, Kirsch GE, Zhang D, Brugada R, Brugada J, Brugada P..
Genetic basis and molecular mechanism for idiopathic ventricular fibrillation..
Nature, (1998), 392 pp. 293-296
[72]
Kim ED, Bischoff FZ, Lipshultz LI, Lamb DJ..
Genetic concerns for the subfertile male in the era of ICSI..
Prenat Diagn, (1998), 18 pp. 1349-1365
[73]
Spahis JK, Wilson GN..
Down syndrome: perinatal complications and counseling experiences in 216 patients..
Am J Med Genet, (1999), 89 pp. 96-99
[74]
Embleton ND, Wyllie JP, Wright MJ, Burn J, Hunter S..
Natural history of trisomy 18..
Arch Dis Child Fetal Neonatal Ed, (1996), 75 pp. F38-F41
[75]
Zergollern L, Hitrec V, Muzinic D..
Clinical and cytogenetic variations of Patau syndrome (demonstration of ten patients)..
J Genet Hum, (1975), 23(Supl.) pp. 135
[76]
Sybert VP..
Cardiovascular malformations and complications in Turner syndrome..
Pediatrics, (1998), 101 pp. E11
[77]
Battaglia A, Carey JC, Cederholm P, Viskochil DH, Brothman AR, Galasso C..
Natural history of Wolf-Hirschhorn syndrome: experience with 15 cases..
Pediatrics, (1999), 103 pp. 830-836
[78]
Doyle EF, Rutkowski M..
Etiology of congenital heart disease..
Cardiovasc Clin, (1970), 2 pp. 1-25
[79]
Wang J, Wilhelmsson H, Graff C, Li H, Oldfors A, Rustin P et al..
Dilated cardiomyopathy and atrioventricular conduction blocks induced by heart-specific inactivation of mitochondrial DNA gene expression..
Nat Genet, (1999), 21 pp. 133-137
[80]
Hamet P, Pausova Z, Adarichev V, Adaricheva K, Tremblay J..
Hypertension: genes and environment..
J Hypertens, (1998), 16 pp. 397-418
[81]
Pratley RE..
Gene-environment interactions in the pathogenesis of type 2 diabetes mellitus: lessons learned from the Pima Indians..
Proc Nutr Soc, (1998), 57 pp. 175-181
[82]
Mechanisms in the pathogenesis of congenital heart defects. En: Pierpont ME, Moller JM, editors. The genetics of cardiovascular disease. Boston: Martinus-Nijhoff, 1985; 3.
[84]
Nora JJ, McGill CW, McNamara DG..
Empiric recurrence risks in common and uncommon congenital heart lesions..
Teratology, (1970), 3 pp. 325-330
[85]
Fearon ER, Vogelstein B..
A genetic model for colorectal tumorigenesis..
Cell, (1990), 61 pp. 759-767
[86]
Faderl S, Talpaz M, Estrov Z, O'Brien S, Kurzrock R, Kantarjian HM..
The biology of chronic myeloid leukemia..
N Engl J Med, (1999), 341 pp. 164-172