Palabras clave
INTRODUCCIÓN
Las enfermedades cardiovasculares y sus complicaciones trombóticas constituyen la causa más frecuente de morbilidad y mortalidad en los países desarrollados del mundo occidental. Se calcula que provocan 16,6 millones de muertes por año, de las cuales casi la mitad son debidas al infarto agudo de miocardio1. En los últimos 20 años se ha observado una estabilización de la mortalidad cardiovascular relacionada con la prolongación de la vida. Sin embargo, en Estados Unidos se ha registrado un incremento entre las mujeres con una muerte cada minuto, lo que representa medio millón de muertes al año y supera las otras 7 causas siguientes de defunción, con un alto impacto socioeconómico debido a la conversión del paciente agudo en crónico2. En los países en vías de desarrollo se aprecia un incremento de su prevalencia; por ejemplo, en Argentina se observa una tasa de mortalidad bruta de 250,6 casos por 100.000 habitantes, lo cual significa 19.000 muertes por infarto agudo de miocardio por año, con mayor incidencia en las grandes ciudades de la región central3.
Los síndromes coronarios agudos (SCA) configuran la expresión más frecuente y actualmente se prefiere su clasificación en síndromes isquémicos sin elevación del segmento ST (angina inestable e infarto de miocardio [IM] sin supradesnivel del segmento ST) y con elevación del segmento ST (IM con supradesnivel del segmento ST)4, basada en el hecho fisiopatológico de la rotura o erosión de la placa con trombo con una obstrucción parcial o total del vaso coronario. Se ha registrado un marcado incremento de los primeros y una estabilización o disminución de los segundos como causa de admisión hospitalaria de los pacientes5.
En los últimos 20 años se ha observado un notable incremento en el conocimiento básico y clínico de la génesis, la progresión y las complicaciones de la enfermedad aterotrombótica, sustrato etiopatogénico de los SCA. El concepto inicial de la rotura o erosión de una placa vulnerable, como causa clínica de los SCA, se ha complicado bastante recientemente. De hecho, hoy día se tiene en cuenta el concepto de «paciente vulnerable o de alto riesgo», que es el que presenta una mayor probabilidad de presentar eventos cardiovasculares en los próximos años. El riesgo de este paciente vulnerable sería el resultado de la suma de la presencia de una placa vulnerable y de una «sangre vulnerable»6. Estudios recientes han demostrado claramente la posibilidad de que un mismo paciente tenga varias placas rotas, aunque sólo una de ellas sea la causante de la manifestación clínica; del mismo modo, se ha establecido la posibilidad de presentar una SCA sin rotura de la placa6,7. En estudios post mortem se ha observado la presencia de ciertos factores cardiovasculares (tabaquismo, diabetes, hiperlipidemia) como causantes de un evento, sin que mediara la rotura de una lesión aterosclerótica6,8.
Paradigma significa un nuevo modelo, mientras que dogma, palabra también derivada del griego, es el fundamento de una doctrina establecida. Estas nuevas evidencias fisiopatológicas que constituyen el primer paradigma tienen importantes influencias en las conductas diagnósticas (segundo paradigma) y en los tratamientos establecidos (tercer paradigma). Probablemente, juntos generen en el futuro el nuevo dogma de los SCA.
PRIMER PARADIGMA: LA NUEVA BIOLOGÍA
Los diferentes aspectos de la fisiopatogenia de la aterotrombosis han evolucionado sustancialmente. En la actualidad se acepta la inclusión de complejos procesos biológicos, como inflamación, apoptosis, la presencia del factor tisular, activadores del sistema inmunológico y otros factores ambientales que, en conjunto, constituyen una verdadera ecuación de variables con un sustrato esencial de isquemia la cual, según las circunstancias, determinará la aparición de un accidente coronario agudo.
La aterosclerosis es una enfermedad sistémica que comienza en la niñez, incluso en la vida prenatal. En estudios recientes se ha demostrado que fetos nonatos ya presentaban lesiones del tipo de las estrías grasas en diferentes territorios vasculares, y que estaban relacionados con el grado de hipercolesterolemia de la madre9. La primera manifestación funcional de alteración arterial es la disfunción endotelial y precede a la primera manifestación anatómica de alteración arterial, que es la estría grasa. En estudios efectuados con IVUS (ultrasonidos intracoronarianos) en corazones de donantes para trasplante cardíaco, con un umbral de 0,5 mm de engrosamiento intimal, se ha hallado una relación directa entre la edad del donante y la incidencia de la enfermedad arteriosclerótica en el corazón donado; así, por ejemplo, la prevalencia de enfermedad coronaria era del 37% en la década de los 20 años, del 71% en la década de los 40 años y > 85% en los mayores de 50 años9,10. La aparición generalizada de pequeñas placas ateromatosas, llamadas «vulnerables» por su aspecto histopatológico o de «alto riesgo» por sus implicaciones pronósticas, es la causa principal, junto con su posterior rotura, de los eventos y las complicaciones en el territorio coronario.
La aterotrombosis es la enfermedad caracterizada por 4 conceptos: es una enfermedad difusa, las lesiones son muy heterogéneas, es más importante la composición de las lesiones que su severidad y, finalmente, es una enfermedad multifactorial.
1. Primer concepto: es una enfermedad difusa porque un subestudio del Framingham de 5.209 pacientes seguidos durante 10 años ha demostrado que los que presentaban un IM tenían un 33% de posibilidades de presentar un accidente cerebrovascular (ACV) o una vasculopatía periférica en los próximos años. De igual modo, un tercio de los pacientes que presentaba un ACV como primera manifestación podría padecer un IM, y viceversa10.
2. Segundo concepto: la aterosclerosis es una enfermedad heterogénea o multiforme, ya que se pueden encontrar al mismo tiempo lesiones arteriales en distintos estadios de evolución en diferentes lechos arteriales de un mismo sujeto. En estudios recientes se ha demostrado con el uso de IVUS que pacientes con SCA tenían múltiples placas inestables y en distintos estadios de evolución11.
3. Tercer concepto: es más importante la composición que la severidad de las lesiones, ya que según un metaanálisis12 con estudios efectuados en pacientes que fallecieron por causa cardiovascular, en el 75% de los casos la lesión causante del acontecimiento podía ser clasificada como «vulnerable»12. Estas placas, que generalmente son excéntricas y producen una estenosis < 50%, tienen un gran contenido lipídico extracelular separado del lumen arterial por una cápsula delgada, y con abundante infiltración de monocitos/macrófagos y linfocitos T que expresan una actividad inflamatoria intensa, especialmente en su hombro. Por el contrario, las placas fibrosas o «estables» son más concéntricas, con núcleos lipídicos intracelulares, sin signos de actividad inflamatoria y cubiertas por gruesas capas de colágeno. Estas placas «vulnerables» modifican el concepto tradicional por este nuevo paradigma y generan la necesidad de nuevos enfoques diagnósticos, ya que la mayoría de estas pequeñas placas inestables son difíciles de detectar o de reconocer mediante la angiografía convencional, considerada anteriormente el «patrón de referencia» por su sensibilidad diagnóstica. En la actualidad se prefiere cambiar la denominación de placa «vulnerable» por la de placa de «alto riesgo» para englobar, de este modo, a todas las lesiones que, sin las características histológicas de las placas vulnerables, también son causantes de lesiones en los territorios carotídeos y vasculares periféricos.
4. Cuarto concepto: se ha involucrado a más de 270 factores reconocidos que participarían en la enfermedad aterosclerótica, entre ellos los factores de riesgo clásicos: hipercolesterolemia, hipertensión arterial, tabaquismo, diabetes, sedentarismo y herencia cardiovascular, y los denominados nuevos factores de riesgo o no tradicionales: hiperhomocisteinemia, lipoproteína Lp(a), agentes infecciosos como Chlamydia pneumoniae, Helicobacter pylori, citomegalovirus y Bacteroides gingivalis, así como la microalbuminuria, los factores inflamatorios (proteína C reactiva [PCR], sustancia amiloidea sérica y recuento de glóbulos blancos) y factores protrombóticos (PAI-1, dímero D, factor de von Willebrand e hiperfibrinogenemia). Todos ellos contribuyen en mayor o menor grado a los cuadros isquémicos agudos y generan el cuarto concepto: la aterosclerosis es una enfermedad poligénica, multifactorial, inflamatoria e inmunológica12-14.
FASES DE LA ATEROTROMBOSIS. EL CAMINO QUE CONDUCE A LOS SINDROMES CORONARIOS AGUDOS
Desde la iniciación de la placa ateromatosa en la primera infancia o en la vida prenatal, su desarrollo hasta la edad adulta y su posible complicación provocada por rotura o erosión de la capa fibrosa, todos los mecanismos fisiopatológicos están basados en la interacción, retroalimentación y potenciación del eje inflamación-trombosis. Su génesis se inicia con la disfunción endotelial, y esta misma disfunción es la que posibilitará su modulación a vulnerable o estable, permitirá su progresión o provocará su regresión, y la protegerá de la rotura o la erosión o la desprotegerá facilitando el accidente isquémico agudo.
Disfunción endotelial. Una ventana a la biología molecular
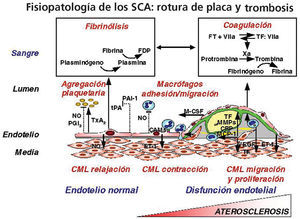
La disfunción endotelial es un fenómeno sistémico, reversible, que se puede considerar como el síntoma patológico inicial del proceso aterosclerótico14,15. El endotelio, cuando funciona normalmente, es un órgano de un trillón de células que producen más de 60 moléculas, las cuales contribuyen a la homeostasis y la hemostasis vascular mediante la regulación de la vasodilatación arterial, la inflamación y proliferación celular, y la modulación de la trombosis-fibrinólisis. En estado disfuncionante se caracteriza por la disminución de la biodisponibilidad de vasodilatadores antiaterogénicos, representada por su molécula principal, el óxido nítrico (NO), que permite la alteración del equilibrio homeostático a favor de los vasoconstrictores proaterogénicos y protrombóticos, como la angiotensina II (A-II). Este desequilibrio conduce a una reducción de la capacidad del endotelio de mantener la homeostasis del medio que se denomina disfunción endotelial, aunque otros autores prefieren nominarla activación endotelial. Esta situación facilita la permeabilidad endotelial para el paso de lípidos, favorece la oxidación de las lipoproteínas, la inflamación, la proliferación de células musculares lisas, la deposición o lisis de la matriz extracelular, la activación plaquetaria y la trombogénesis16. Por otra parte, el endotelio también regula, como se ha mencionado con anterioridad, la producción de factores trombóticos y antitrombóticos, fibrinolíticos y antifibrinolíticos, factores de crecimiento, proteínas inmunogénicas y sustancias proinflamatorias (fig. 1).

Fig. 1. Mecanismos de homeostasis presentes en el endotelio normal y su alteración en la activación o disfunción endotelial13. CAM: moléculas de adhesión celular; CML: células musculares lisas; ET-1: endotelina 1; FT: factor tisular; M-CSF: factor estimulante del macrófago; MCP-1: proteína quimiestática de monocitos 1; MMP: metaloproteinasas matriciales; NO: óxido nítrico; PAI-1: inhibidor 1 del t-PA; PGI2: prostaciclina; t-PA: activador tisular del plasminógeno; TxA2: tromboxano A2; VEGF: factor de crecimiento del endotelio vascular.
Las placas ateromatosas tienden a localizarse en las bifurcaciones arteriales, y esta selectividad señala la importancia de las condiciones reológicas del flujo sanguíneo en la determinación del lugar donde se ubica la placa. Más aún, la expresión genética de las células endoteliales es modulada por los cambios agudos de las condiciones de flujo17.
Un endotelio disfuncionante genera un entorno proaterogénico caracterizado por inflamación, proliferación y estado protrombótico que favorecen la instalación de la aterosclerosis17. Además, los mediadores derivados de las plaquetas, como la serotonina, inducen vasoconstricción en presencia de un endotelio activado18,19, y la respuesta vasoconstrictora es magnificada por la liberación de endotelina 120, el más poderoso de los vasoconstrictores de la economía, cuya concentración se encuentra siempre elevada en el plasma de los pacientes ancianos con aterosclerosis avanzada y en las lesiones coronarias causantes21,22.
La disfunción endotelial está involucrada en el reclutamiento de células inflamatorias dentro de la íntima arterial y la iniciación del proceso aterosclerótico, para lo cual el endotelio expresa moléculas de adhesión celular como selectinas (moléculas de adhesión vascular celular [V-CAM] e intercelular [I-CAM]), y sintetiza y libera citocinas inflamatorias y proteínas quimiotácticas que contribuyen a la migración y penetración de monocitos y linfocitos T en la pared arterial. Los monocitos instalados en el subendotelio se activan y transforman en macrófagos que retroalimentan la inflamación y producen quimiotactinas, que continúan reclutando nuevos monocitos. Paralelamente, las células musculares lisas se modulan a secretoras y generan colágeno y proteoglucanos que construirán la capa fibrosa23.
Los factores de riesgo clásicos (hipercolesterolemia, diabetes, hipertensión arterial, tabaquismo, sedentarismo, obesidad, etc.) y algunos de los llamados nuevos factores de riesgo (hiperhomocisteinemia, depresión, infecciones, etc.) tienen todos el común denominador de provocar una sobreproducción de especies reactivas de oxígeno que generan un estado de «estrés oxidativo»24. Este estado, directamente o a través de las proteínas calentadoras-60 (Heat Shock Protein-60), bloquea el inhibidor del factor nuclear kappa-beta (I-κβ) permitiendo su replicación. El factor nuclear kappa-beta (NF-κβ) es un factor de transcripción que regula varias decenas de genes involucrados en la inflamación que liberan diversas citocinas proinflamatorias, como el factor de necrosis tumoral alfa (TNF-α), las interleucinas (IL) IL-1, IL-6, moléculas de adhesión leucocitarias, quimocinas y quimiotactinas. Varias de estas citocinas provocan inhibición de la actividad de la óxido nítrico sintetasa constitutiva endotelial (ONS-III), lo que reduce la biodisponibilidad de NO, mientras favorecen la síntesis y la actividad de la A-II, lo que promueve una franca reacción inflamatoria con el consiguiente daño celular20-24. A su vez, muchas de las citocinas proinflamatorias inducen la replicación del NF-κβ y, de esta manera, retroalimentan el circuito inflamatorio25. Por estas razones, el estrés oxidativo es considerado como el disparador inicial que induce disfunción endotelial, y es el mecanismo patogénico común que relaciona los efectos de los factores de riesgo sobre el endotelio.
Recientemente, el grupo de Hill et al26 comunicó el valor de las denominadas células endoteliales progenitoras. Este tipo de «células madre» o stem cell de médula ósea tiene la capacidad de migrar desde su origen y regenerar las células endoteliales de los vasos del organismo durante toda la vida. La concentración plasmática de estas células disminuye con la edad, y su número en el plasma se relaciona con la respuesta vasodilatadora dependiente del endotelio y con la presencia de factores de riesgo asociados. Su cuantificación en el plasma guarda una relación inversa con el promedio de riesgo de Framingham, reduciéndose en los pacientes con riesgo intermedio-alto. Esto explica la disminución presente en pacientes con SCA, en los cuales su número desciende a valores mínimos; esto expresa la pérdida del poder regenerador del endotelio y favorece los fenómenos de apoptosis y erosión endotelial.
En esta etapa inicial se reconocen 3 niveles de alteración endotelial: a) la activación endotelial, fenómeno inicial que ejercita el endotelio diariamente para mantener la homeostasis de las múltiples funciones que regula; b) la disfunción endotelial leve, por la cual los múltiples factores de riesgo alteran la regulación del endotelio y comienzan a primar acciones de aumento de producción de moléculas de adhesión y proinflamatorios con la producción elevada de citocinas; este estadio es potencialmente reversible con fármacos protectores del endotelio, y por último c) la fase de disfunción endotelial avanzada, con profundas alteraciones anatómicas y funcionales que conducen a mecanismos inflamatorios y protrombóticos. Además, al constituir una etapa más terminal, la respuesta terapéutica no siempre es efectiva. En todas estas etapas, el denominador común y sustrato fisiopatológico presente en todas ellas es la endotelitis, que progresa desde las fases iniciales a las avanzadas y establece el puente con la trombosis, factor que hace evolucionar a saltos la entidad aterosclerótica y conduce a sus temidas complicaciones, como los SCA14. Nuestro grupo observó que la respuesta vasodilatadora dependiente del endotelio, valorada mediante el test de isquemia braquial en pacientes portadores de angina inestable, es prácticamente nula comparada con la de los pacientes con factores de riesgo clásicos. Se postuló el concepto de «endotelio atontado» para explicar la baja respuesta vasodilatadora en los cuadros agudos, en comparación con el «endotelio hibernado», que es el que se cuantifica en presencia de factores de riesgo y, de este modo, genera un factor facilitador de la rotura de una placa de alto riesgo que desencadena un accidente isquémico agudo27 (fig. 2).

Fig. 2. Función endotelial alterada en la angina inestable. Endotelio «atontado», lo que demuestra una respuesta vasodilatadora fuertemente deprimida comparada con la expresada en presencia de factores de riesgo27. AI: angina inestable; Col: colesterol; FR: factores de riesgo.
El modelo unificado propuesto a fines de la década pasada tiene plena vigencia en la actualidad. Éste sostiene que los múltiples factores de riesgo generan estrés oxidativo y por este mecanismo conducen a la disfunción endotelial en toda su magnitud. En los estadios avanzados predominan los mecanismos de vasoconstricción, proliferación celular, inflamación y trombosis. Todos ellos actúan según la denominada teoría de la «respuesta a la lesión»28. La disfunción endotelial, no sólo contribuye a la instalación de la ate-roesclerosis, sino que es la causante de su progresión, del control de su regresión, de la falta de protección de las placas de alto riesgo que permite su rotura, de la instalación del trombo obstructivo u oclusivo, y hasta de su posible fibrinólisis, por lo que es el factor más importante de todas las etapas de la enfermedad aterotrombótica.
Eje inflamación-inmunidad y aterotrombosis
Hay un creciente número de investigaciones básicas y clínicas que relacionan la inflamación y la aterotrombosis28-30. La aterogénesis se inicia en áreas vasculares donde las células endoteliales comienzan a expresar moléculas de adhesión celular selectivas en su superficie que involucran diferentes clases de leucocitos. En particular, la VCAM-1 liga precisamente los monocitos y los linfocitos T, que son los que se encuentran en el inicio del desarrollo del ateroma experimental. Los ratones manipulados genéticamente para no producir VCAM-1 retrasan o detienen el desarrollo de las placas ateromatosas29,30.
Al principio, los leucocitos ruedan sobre la superficie endotelial, luego se adhieren y posteriormente penetran en la íntima por los espacios intercelulares, atraídos por proteínas quimiotácticas causantes de esta migración. Éste es el caso de la proteína quimiotáctica de monocitos 1 (MCP-1), que es la encargada de la atracción y migración de los monocitos hacia los sitios donde se está formando la lesión inicial del ateroma31. Una vez adheridos e introducidos en la íntima por acción de las VCAM-1 e ICAM-1, los leucocitos no sólo participan del proceso inflamatorio, sino que lo retroalimentan y perpetúan. Los macrófagos expresan receptores «carroñeros» (scavenger receptors) que fagocitan las lipoproteínas especialmente modificadas por la oxidación (LDL oxidadas), lo que genera las células espumosas (foam cells).
El proceso inflamatorio no sólo es el promotor de la iniciación y progresión de la ateroesclerosis, sino que contribuye decisivamente a la precipitación de las complicaciones trombóticas. El macrófago activado, muy abundante en el ateroma, puede producir enzimas proteolíticas, como las metaloproteinasas matriciales (MMP), familia de más de 20 componentes que incluye colagenasas, gelatinasas y elastasas, capaces de degradar los componentes de la capa fibrosa protectora del ateroma, adelgazándola, debilitándola y haciéndola suceptible a la rotura. Se ha comunicado32 la presencia de 3 metaloproteinasas (colagenasas) intersticiales en el ateroma, MMP-1, 8 y 13.
Las células musculares lisas de la pared arterial migran alrededor del núcleo lipídico y se modulan fenotípicamente de contráctiles a secretoras, produciendo colágeno y proteoglucanos que conformarán la capa fibrosa de la placa. Los linfocitos T liberan interferón gamma en el seno de la placa que inhibe la síntesis de colágeno por las células musculares lisas de la pared arterial, lo que limita su capacidad de renovar el colágeno que refuerza la cubierta de la placa33. Por otra parte, el interferón gamma activa la producción de MMP por parte de los macrófagos.
Todas estas células que se encuentran en la pared arterial, endoteliales, musculares lisas, macrófagos y linfocitos T, una vez activadas son capaces de producir factor tisular, que es el principal disparador del proceso de coagulación y, por tanto, uno de los factores primordiales de la trombogénesis de la placa. Los mediadores inflamatorios modulan la expresión del factor tisular por los macrófagos activados, lo cual demuestra una relación comprobable entre inflamación y trombosis33.
Los marcadores serológicos de inflamación, como la PCR34,35, la IL-635, la sustancia amiloide sérica A36, la ICAM-1 soluble37, y el CD40L38, acompañan con frecuencia a las diversas manifestaciones de enfermedad coronaria. En múltiples investigaciones multicéntricas, el incremento de sus concentraciones séricas se correlacionó con un pronóstico adverso, que refleja la contribución de la inflamación como instigadora de la inestabilidad de la placa aterosclerótica.
En opinión de Libby et al39, esta representación multifactorial de la aterogénesis va de un extremo del universo representada por la hipercolesterolemia familiar, donde en su génesis contribuye fuertemente el colesterol unido a lipoproteínas de baja densidad (cLDL), al otro extremo representado por la arteriopatía postrasplante cardíaco, donde el mecanismo principal es una fuerte activación del sistema inmunológico. En el medio de este horizonte, la aterosclerosis habitual es una entidad mixta entre los factores clásicos y los factores con estímulo inmunogénico. Las placas ateroescleróticas en desarrollo están infiltradas no sólo de macrófagos, sino de linfocitos T(CD4)Th, llamados helper o de ayuda, y linfocitos T (CD8), lo que sugiere fuertemente una respuesta inmunológica específica. Sin embargo, aún no hay acuerdo entre los investigadores respecto a la atribución de esa respuesta a un protagonismo protector o deletéreo en el desarrollo de la placa. En efecto, los linfocitos T(CD4)Th1 producen TNF-α, interferón gamma e IL-2, todas sustancias proinflamatorias que activan los macrófagos y causan las reacciones de hipersensibilidad tardía. Por el contrario, los linfocitos-T(CD4)Th2 generan IL-4, IL-5, IL-10 e IL-13, todas ellas moléculas antiinflamatorias que promueven respuestas por anticuerpos e inhiben enérgicamente la actividad de los macrófagos40,41.
En las placas ateroscleróticas de animales de experimentación y en humanos se ha podido determinar la presencia de citocinas inflamatorias producidas por la variante de linfocitos T(CD4)Th1, como interferón gamma e IL-12, con un microambiente proinflamatorio semejante al de la artritis reumatoidea42-46. En cambio, en otros estudios no se las pudo detectar, lo que hizo sospechar una reducción de la actividad inflamatoria, por lo que se cree que el equilibrio entre las variantes de linfocitos T(CD4) Th1 y Th2 puede desempeñar un papel primordial en la progresión o regresión de la placa. En este sentido, se ha demostrado que las estatinas desempeñan una actividad inmunomoduladora47,48.
POLIMORFISMOS GENÉTICOS, INFLAMACIÓN Y ROTURA DE LA PLACA
Recientes publicaciones de Maseri et al49 encuadrarían el complejo mosaico del determinismo que llevan los SCA. En un extremo, se encuentra un paciente joven sin factores de riesgo, con una puntuación de Framingham de bajo riesgo, una dieta y un ejercicio regular apropiados y que desarrolla un IM, con una angiografía posterior que revela múltiples lesiones coronarias que requieren cirugía de revascularización miocárdica. En el otro extremo del espectro se encuentra un paciente de edad avanzada con múltiples factores de riesgo, una puntuación de Framingham de alto riesgo, obeso, sedentario y diabético. Este paciente, paradójicamente, se encuentra asintomático a los 92 años de edad. Estos ejemplos límite representan los extremos de un universo complejo, donde múltiples factores clásicos, noveles y genéticos interactúan de manera muy variada. Recientemente se han descubierto numerosos polimorfismos genéticos. La mutación del gen MEF2A presente en el cromosoma 13 que codifica numerosos factores de transcripción (principalmente el factor de incremento monocítico 2) se asocia en un 100% al incremento del riesgo de IM y ACV50. Además, variantes del gen ALOX5AP, que codifica la proteína activadora de la lipooxigenasa 5, indispensable en la regulación inflamatoria de los leucotrienos, se asocia fuertemente a un doble riesgo de ACV e IM.
Este gen fue codificado por el grupo de CODE, que lo detectó en el cromosoma 13q12-13 mediante un amplio mapeo de genes en pacientes con IM en Islandia51. Esto genera una nueva y prometedora ventana terapéutica, que se encuentra en fase 2 de desarrollo, con el fármaco DG 031, empleado anteriormente para el asma, como inhibidor de los leucotrienos mediante el procedimiento denominado farmacogenoma.
Recientemente, investigadores japoneses han encontrado una asociación significativa entre el gen que codifica la galectina-2 (LGALS2), proteína vinculada a moléculas mediadoras de inflamación como la linfotoxina alfa, con pacientes que previamente habían presentado un IM. En este estudio de casos y controles en el que se incluyó a 2.638 pacientes con IM y 2.038 controles, los portadores de este polimorfismo presentaban un mayor número de fenómenos de rotura de placa y posterior SCA52.
PLACA VULNERABLE O DE ALTO RIESGO
Entre la estría grasa y la placa vulnerable se encuentra el estadio intermedio de la evolución de la aterosclerosis, constituido por la aparición de placas de ateroma. En este transcurso, la clasificación de la American Heart Association propone 5 fases de evolución; la fase 2 se expresa con la presencia de placas tipo IV y V.
Estas placas, que histopatológicamente corresponden a placas vulnerables, durante su evolución pueden migrar hacia la fase 3, con la progresión y consolidación de la placa de ateroma, y se expresan clínicamente como una progresión del grado de angina. También pueden evolucionar hacia la fase 4, con la rotura o erosión, y producir un trombo parcial o total que desencadena un síndrome isquémico agudo53 (fig. 3).

Fig. 3. Placas tipo IV y V de la clasificación de la American Heart Association con rotura y complicaciones isquémicas52.
Estas placas de tipo IV y Va pueden tener distinta expresión fenotípica y manifestarse con diversa morfología, con rotura, fisura-cicatrización y hemorragia intraplaca (que representan el 70% de las placas presentes en los SCA y con preferencia en los varones). Con menos frecuencia se presenta como una erosión (usualmente en mujeres fumadoras, hipercolesterolémicas o diabéticas) o con calcificación de la placa54.
Naghavi6,8 propone una serie de criterios mayores y menores con implicaciones diagnósticas y pronósticas. Los criterios mayores de placas de alto riesgo son: inflamación en el hombro de la placa, núcleo lipídico grande con cápsula delgada, denudación endotelial con agregación plaquetaria y placa con fisura o rotura superficial. Los criterios menores son: nódulo superficial calcificado, color amarillo brillante, hemorragia intraplaca, estenosis crítica y remodelado positivo. Con la presencia de 2 criterios mayores o 1 mayor y 2 menores se confirma el diagnóstico.
El riesgo de rotura de una placa depende de la vulnerabilidad intrínseca y del estrés mecánico a que es sometida. Los determinantes de vulnerabilidad pueden clasificarse en extrínsecos o intrínsecos (tabla 1). Los extrínsecos están principalmente en conexión con su localización, mientras que los intrínsecos se relacionan con la composición de las lesiones y tienen mayor importancia en la patogenia de la enfermedad, ya que es posible interferirlos mediante tratamiento dietético y/o farmacológico14.

Tres propiedades intrínsecas de la placa determinan su vulnerabilidad: a) el tamaño y la consistencia del núcleo ateromatoso; b) la estructura y la firmeza de la capa fibrosa, y c) el proceso inflamatorio dependiente de los monocitos-macrófagos activados14.
Los trombos se forman sobre placas ateroescleróticas rotas y ricas en lípidos32,33, pero también pueden generarse por la simple erosión de la superficie endotelial, como sucede en los pacientes con factores de riesgo protrombóticos (tabaquismo, diabetes, hipercolesterolemia, etc.)55. Se calcula que los trombos se forman por la rotura de placas entre 1,3 y 3 veces más que por erosión endotelial56.
La trombosis consecuente a la rotura de placa se suele observar en las que tienen un bajo grado de obstrucción, que habitualmente pueden no ser evidenciables mediante coronariografía. La trombosis por erosión endotelial es más frecuente en los sitios con altos grados de estenosis y se ha comunicado que es más común en mujeres y en varones jóvenes con factores de riesgo56,57.
La erosión endotelial expone el colágeno subyacente a la sangre circulante, lo que promueve la trombosis. Pequeñas áreas de erosión se acompañan de trombos plaquetarios microscópicos sin manifestación clínica, aunque pueden estimular la proliferación de las células musculares lisas o bien, una vez regenerado el endotelio, alterar su respuesta vasomotora. Áreas mayores de erosión, especialmente en presencia de factores de riesgo, pueden facilitar la constitución de trombos plaquetarios de mayores dimensiones, con contenido de fibrina y hematíes, los cuales pueden obstruir u ocluir el vaso. Esta última forma de trombosis se ha asociado a una marcada acumulación de macrófagos y células espumosas en el subendotelio, un incremento en el número de linfocitos T y la expresión de complejos mayores de histocompatibilidad antigénica tipo II por las células musculares lisas, lo cual conduce a la suposición de que la erosión endotelial es la consecuencia de la actividad inflamatoria58,59. No obstante, la patogenia puede ser distinta en las mujeres y se ha observado en placas ricas en células musculares lisas y proteoglucanos pero pobre en lípidos, macrófagos y células inflamatorias60.
SANGRE VULNERABLE Y PACIENTE VULNERABLE
En un tercio de los accidentes coronarios agudos, especialmente en los casos de muerte súbita, no se ha encontrado rotura de placas con núcleos lipídicos manifiestos, es decir, de tipo vulnerable, sino placas con una marcada estenosis fibrótica y con sólo una erosión superficial con pérdida del endotelio61,62. En esos casos, la formación del trombo oclusivo depende más de un estado de hipertrombogenia generado por factores sistémicos. En realidad, estos factores sistémicos, incluida la elevación de los niveles séricos del cLDL, el hábito tabáquico, la hiperglucemia, la hemostasia y otros, están asociados con un incremento de la trombogenicidad de la sangre63.
Se ha observado que el aumento de los valores séricos del cLDL incrementa la trombogenicidad de la sangre y el crecimiento del trombo en condiciones reológicas definidas64,65. La disminución de la concentración de cLDL con estatinas ha mostrado un descenso del crecimiento del trombo de aproximadamente un 20%65. El interrogante que surge es cuán intenso es este efecto antitrombótico, como por ejemplo bajo el efecto de las estatinas, documentado en grandes ensayos prospectivos, y con qué magnitud contribuye a la reducción total de accidentes vasculares, incluidos la muerte súbita, los eventos coronarios y los ACV66,67.
El tabaco aumenta la actividad nerviosa simpática y, por tanto, la liberación de catecolaminas68, lo cual potencia la activación plaquetaria e incrementa los valores séricos de fibrinógeno. Los efectos dependientes de las catecolaminas en la circulación de la sangre pueden explicar, no sólo el aumento de la incidencia de muerte súbita y accidentes vasculares agudos después del estrés emocional o físico, sino también la distribución circadiana de estos eventos69.
Los pacientes diabéticos, en especial los que tienen un control ineficiente de la enfermedad, presentan un incremento de la trombogenicidad de la sangre70. Las plaquetas de los pacientes con diabetes tienen un aumento de la reactividad y la agregabilidad, y exponen una variedad de proteínas de adhesión dependientes de la activación71. La función plaquetaria anormal está reflejada en el incremento del consumo de plaquetas y el aumento de la acumulación de plaquetas en los vasos, con sus paredes alteradas por la aterosclerosis.
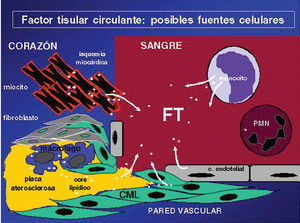
Observaciones recientes indican que los estados de hipertrombogenia asociados con un cLDL elevado, el hábito tabáquico y la diabetes comparten un mecanismo común de activación de la interacción leucocitos-plaquetas asociado a la liberación de factor tisular y la activación de la trombina. Específicamente, en la sangre de los pacientes con diabetes mellitus se observa un aumento de la agregación leucocitaria y plaquetaria circulante. El estado protrombótico de la diabetes en presencia de microalbuminuria diabética está también asociado a un incremento de la expresión de la actividad procoagulante de los monocitos. El aumento de la actividad procoagulante de la diabetes se atribuye a los leucocitos, que en parte activan el mecanismo del factor tisular y contribuyen a la alta trombogenicidad de los pacientes diabéticos71-73 (fig. 4).

Fig. 4. Fuentes de síntesis del factor tisular79. CML: célula muscular lisa; FT: factor tisular.
Estudios recientes han encontrado un aumento de las concentraciones de antígenos del factor tisular circulante en pacientes con enfermedad coronaria74, y el factor tisular circulante se ha asociado a un aumento de la trombogenicidad de la sangre en pacientes con angina inestable y enfermedad coronaria crónica56,75. Los niveles de factor tisular en la sangre circulante también se han considerado predictores de la evolución en pacientes con angina inestable75,76.
Como se ha comentado con anterioridad, las placas ateromatosas ricas en lípidos contienen macrófagos y factor tisular dentro del núcleo lipídico, que podrían ser los causantes, en gran parte, de la alta trombogenicidad de este tipo de lesiones77. Además, el factor tisular fue identificado dentro de los trombos formados en la luz de las arterias coronarias. Por otra parte, la inhibición específica del factor tisular reduce significativamente la trombogenicidad de la placa78. El inhibidor específico del factor tisular recombinante (tissue factor pathway inhibitor [TFPI]) está usualmente expresado en la adventicia de la pared de las grandes arterias y, dentro de los vasos con ateromatosis, en los macrófagos y en áreas necróticas de la placa. La producción local de TFPI podría regular la actividad procoagulante y los accidentes trombóticos dentro de las placas ateromatosas78,79.
Además de los restos de macrófagos apoptóticos y de las micropartículas de las placas ateroesclerosas, parece que los monocitos activados en la sangre circulante son la fuente de micropartículas de factor tisular, y podrían ser el resultado de la activación de los factores de riesgo mencionados anteriormente y de otros, contribuyendo a los eventos trombóticos. Dentro del contexto de los posibles efectos proinflamatorios y protrombóticos de la sangre circulante con altos valores de cLDL, tabaquismo y diabetes, hay una creciente evidencia de que los monocitos circulantes en mayor grado y los leucocitos, en general en menor cuantía, pueden estar involucrados en la producción de factor tisular y trombogenicidad79. Recientemente, Hutter et al79 demostraron en ratones Apo E que había una relación directa entre la concentración de caspasa 3, factor tisular y CD 68 como expresión de la apoptosis o suicidio celular de los macrófagos, expuestos a un estímulo de hipercolesterolemia, en placas ricas en lípidos comparadas con placas fibrosas, elemento que sería un factor determinante en la síntesis de factor tisular79.
PARADIGMA DIAGNÓSTICO. HACIA UNA ESTRATEGIA CON MÚLTIPLES BIOMARCADORES
Se ha descrito que los SCA son un complejo síndrome con múltiples causas, análogo a la anemia o la hipertensión arterial. El grupo TIMI describió las 5 causas que llevan a los SCA: a) rotura de placa con trombosis; b) obstrucción progresiva mecánica; c) inflamación; d) angina inestable secundaria, y e) obstrucción dinámica. Actualmente, los avances en la comprensión de la fisiopatología y las consecuencias de los SCA generan la necesidad del empleo de una estrategia de multibiomarcadores80: marcadores de necrosis miocítica (troponina, isoenzima MB de la creatincinasa, mioglobina), marcadores de inflamación (PCR, CD40L, SAA), marcadores de daño vascular (microalbuminuria, aclaramiento de creatinina), marcadores de aterosclerosis acelerada (glucosa plasmática, HbA1C) y marcadores de estrés hemodinámico (péptido natriurético cerebral [BNP], NT-proBNP). Todos ellos llevan información de los procesos en juego predominantes en la fisiopatología de este mapa en el espectro de los SCA y permitirían orientar el tratamiento hacia el eje de mayor preponderancia determinante en cada caso. Su asociación daría una información más completa respecto del riesgo isquémico y los eventos coronarios mayores. Un ejemplo de ello es el subestudio GUSTO-IV, donde el BNP o su precursor NT-proBNP asociado a marcadores de daño vascular (aclaramiento de creatinina), de necrosis miocárdica (troponina T o I) o de inflamación (PCR, CD-40L) incrementan el riesgo de mortalidad hasta 25 veces.
La PCR, lo mismo que el fibrinógeno y la sustancia amiloide A, son reactantes de fase aguda y marcadores sensibles de inflamación. Su elevación en el plasma constituye el segundo paradigma de esta entidad. Se ha comunicado que el aumento de los valores séricos de PCR es un buen predictor de eventos coronarios agudos35, y puede ser también un marcador de pronóstico útil en la predicción de eventos trombóticos. Aún hay dudas acerca de si la PCR refleja el componente inflamatorio de la placa aterosclerótica o de la sangre circulante, y si es un marcador clínico o un elemento biológico activo en la formación de trombos en el sitio de la placa ateromatosa rota o dañada81. No obstante, estudios recientes apoyan la hipótesis de que es un activador del monocito circulante y de las células endoteliales de la pared arterial82.
Numerosos trabajos de Ridker et al83, e incluso un metaanálisis con 19 trabajos científicos, efectuados tanto en varones como en mujeres, demuestran que la PCR ultrasensible predice eventos cardiovasculares mayores e incrementa 2-3 veces su prevalencia. Además, recientemente, el mismo grupo ha encontrado un mayor valor predictivo con concentraciones mayores que los habituales de 3 mg/l. Los pacientes que superan registros de PCR de 10 y hasta 20 mg/l tienen un incremento de 4 a 5 veces e incluso un mayor valor predictivo en la puntuación de riesgo de Framingham, en la población de riesgo intermedio84. Sin embargo, los grupos de Pepys et al85 y Danesh et al86, en un análisis del trabajo efectuado en Islandia y en un reciente metaanálisis de su grupo, solamente encuentran en la PCR un modesto valor predictivo de eventos e, incluso, cuestionan las guías del CDC/AHA en la indicación de su utilización en la población de riesgo intermedio85-87.
CONCLUSIÓN
Los SCA son la última etapa de un conjunto de múltiples factores que llevan a su complicación final: la rotura de una placa vulnerable o la erosión con la trombosis subsecuente. Los procesos de disfunción endotelial, inflamación sistémica e intraplaca, la función del sistema inmunológico activado representado por el macrófago y los linfocitos T y B, principalmente, la apoptosis celular, y la inducción por ésta de producción aumentada del factor tisular con el desarrollo de la sangre vulnerable, son algunos de los múltiples factores involucrados en la aterotrombosis, y que llevan finalmente hacia la isquemia o necrosis miocárdica.
Este nuevo paradigma brindado por este novel escenario nos lleva a la necesidad de generar un nuevo marco en el terreno diagnóstico (segundo paradigma), donde probablemente la estrategia de multibiomarcadores sea la más apropiada por el amplio espectro de factores en juego, y nos conduzca a una nueva estrategia terapéutica (tercer paradigma) con el uso de terapia combinada y orientada según el sustrato fisiopatológico sugerido por dichos marcadores. Todo esto nos lleva a un nuevo horizonte futuro, el que produce este nuevo dogma o doctrina desarrollada por este nuevo camino, donde la genética y la biología molecular son 2 de sus principales actores.
Correspondencia: Dr. J.O. Vilariño.
Hospital Alejandro Gutiérrez. Casey 632. Venado Tuerto (2600).
Santa Fe. Argentina.
Correo electrónico: jorvil@waycom.com.ar