Palabras clave
INTRODUCCIÓN
En los últimos 20 años la genética ha ido tomando un papel cada vez más importante en el progreso de las ciencias médicas. Éste se ha notado especialmente en la medicina cardiovascular, lo que ha llevado a la creación del término cardiogenética. La cardiología ha incorporado progresivamente los nuevos avances de la biología molecular y la genética, lo que ha permitido conocer en pocos años numerosos genes causantes de enfermedades cardiacas. Existen múltiples afecciones cardiacas genéticamente determinadas, con o sin cardiopatía estructural acompañante, que pueden predisponer a la aparición de arritmias y de muerte súbita1,2. Estas enfermedades son producto de la alteración en la codificación genética de tres grandes familias de proteínas:
1. Proteínas sarcoméricas, que se encargan de generar la fuerza en el miocito cardiaco y están en la causa de la miocardiopatía hipertrófica (MCH)3.
2. Proteínas del citoesqueleto, que están encargadas de la transmisión de esa fuerza a las células colindantes y causan la miocardiopatía dilatada (MCD)4.
3. Proteínas que codifican para los canales iónicos, encargadas de mantener el equilibrio iónico intracelular y extracelular y causa de las arritmias familiares5.
A pesar de todos los avances conseguidos, nada es tan sencillo como parece, ya que hay un solapamiento importante entre las enfermedades y los genes6. Por ejemplo, la troponina T puede causar tanto MCD como MCH7; el canal de sodio SCN5A es uno de los genes que originan el síndrome de Brugada, el síndrome de QT largo tipo 3 (SQTL3) y también de alteraciones familiares de la conducción8,9. La integración de estas alteraciones genéticas nos permitirá obtener una información clave de la estratificación del riesgo de muerte súbita que conllevan algunas de estas enfermedades.
GENÉTICA
Todo organismo vivo tiene su propia información genética contenida en la molécula de ADN. En ella se encuentran las unidades de herencia, los genes. Los humanos tenemos 70.000 genes distribuidos en 23 pares de cromosomas localizados en el núcleo de las células (22 autosómicos y 1 par sexual) y un único cromosoma mitocondrial. Cada par de cromosomas (homólogo) tiene los mismos genes —de los que tenemos dos copias, denominadas alelos— (fig. 1)10. Cada gen contiene la información necesaria para la síntesis de una proteína, la unidad funcional del organismo, ya que el buen funcionamiento de éste está basado en la síntesis perfecta de todas las proteínas necesarias.

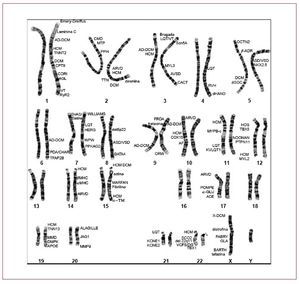
Fig. 1. Cariotipo humano normal en el que se indican, entre otros, los genes que pueden afectarse en diferentes miocardiopatías y su localización en cada uno de los cromosomas10.
La molécula de ADN está formada por cuatro tipos distintos de nucleótidos, repetidos millones de veces. Cada grupo de tres nucleótidos codifica un determinado aminoácido. Éste es un proceso en cadena; los primeros tres nucleótidos codifican el primer aminoácido y los tres siguientes el segundo, y así consecutivamente. Esta síntesis está determinada por el orden que sigue la secuencia de nucleótidos en el ADN. La acumulación progresiva de todos los aminoácidos del gen da lugar a la creación de la proteína. En el genoma humano se ha descrito que menos del 1,5% de éste contiene secuencias que codifican para proteínas11. El resto es material que puede ayudar al correcto funcionamiento, pero su función no está claramente establecida aún.
En ocasiones se puede producir la inserción, deleción o el cambio en el orden de los nucleótidos. Son defectos genéticos denominados mutaciones (fig. 1); éstos pueden producir la síntesis de una proteína diferente o defectuosa y ocasionar una enfermedad. Que el individuo desarrolle o no una enfermedad a causa de la mutación depende de la importancia de la proteína en la función general del cuerpo humano. Si la mutación afecta al ADN de una célula germinal o reproductiva, se transmitirá a las siguientes generaciones y causará una enfermedad hereditaria.
Las enfermedades hereditarias se clasifican en:
1. Alteraciones cromosómicas, con la deleción o adición de una parte o un cromosoma entero.
2. Enfermedades poligénicas (las más frecuentes), que se deben a la interacción de diferentes genes.
3. Enfermedades monogénicas. Sólo un gen causa la enfermedad, y su patrón de herencia sigue las leyes de Mendel:
- Enfermedades autosómicas dominantes. Uno de los alelos heredados es defectuoso y el otro es normal. El carácter dominante de la enfermedad significa que con un solo alelo afectado por una mutación ya se puede originar la enfermedad. La descendencia tiene un 50% de posibilidades de ser portadores de esta enfermedad si uno de los padres está afecto. Cada generación tiene afectados y tanto los varones como las mujeres pueden heredar y transmitir la enfermedad.
- Enfermedad autosómica recesiva. Requiere que los dos alelos sean defectuosos para que se produzca la enfermedad. Por lo tanto, es una forma menos común que la autosómica dominante. Si ambos padres son portadores, la descendencia tiene un 25% de posibilidades de padecer la enfermedad, y un 50% de posibilidades de ser portadores.
- Enfermedades ligadas al sexo. Las mujeres son las portadoras en uno de los cromosomas X. Los varones, al poseer un solo cromosoma X, padecen la enfermedad si heredan el cromosoma mutado.
- Enfermedades mitocondriales. Siempre son transmitidas por la mujer porque el cromosoma mitocondrial proviene siempre de la madre. Por lo tanto, no se produce transmisión de varón a varón.
En general, todos los seres humanos presentan pequeñas variaciones en un lugar determinado del ADN, denominadas polimorfismos. A diferencia de las mutaciones, los polimorfismos carecen de magnitud para producir enfermedades, pero pueden alterar la respuesta del individuo a ellas y producir variaciones en la predisposición, la evolución y la respuesta al tratamiento12,13.
ARRITMIAS
Las arritmias cardiacas son una de las mayores causas de mortalidad y morbilidad en la población, especialmente en los pacientes con enfermedad cardiaca previa14, y causan cada año casi 1 millón de casos de síncope y muerte súbita en Europa y América15. Las arritmias, como todas las enfermedades, resultan de la interacción de factores ambientales y genéticos (fig. 2)12. En las últimas décadas, numerosos estudios han centrado sus hipótesis en los factores arritmogénicos ambientales —tanto estructurales como funcionales—, además de los factores étnicos16. Se ha visto que algunas de las complicaciones en las arritmias sólo se presentan cuando existe una interacción perfecta entre los diferentes factores. Los mecanismos básicos que predisponen a las arritmias en los pacientes, con y sin enfermedad cardiaca previa, no han sido aún bien definidos, aunque se han identificado mutaciones en genes que codifican para canales iónicos cardiacos, como factores de riesgo en la patogenia de arritmias letales y no letales17,18.

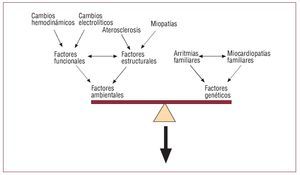
Fig. 2. La arritmogénesis depende de la interacción de diferentes factores, tanto ambientales como genéticos. El equilibrio entre estos factores evita la formación de arritmias. Un factor ambiental predisponente o un factor genético previo pueden ser suficientes para crear el sustrato necesario para la arritmogénesis12.
Las arritmias familiares pueden clasificarse en dos grandes grupos dependiendo de su asociación con otras enfermedades cardiacas19: a) causa primaria, en la que la base de la arritmia se presenta las propiedades eléctricas del corazón20, y b) causa secundaria, en la que la arritmia se debe a una miocardiopatía familiar de base, caracterizada por hipertrofia3, dilatación4 o infiltración fibroadiposa21. Si bien el mecanismo de ambos grupos es diferente, la muerte súbita es la característica común de todas estas enfermedades devastadoras22.
CANALOPATÍAS
Los canales iónicos son proteínas transmembrana que permiten el paso de iones dentro y fuera del miocardiocito; este proceso se rige por una sincronización entre la apertura y el cierre de los canales a raíz de un gradiente eléctrico que origina el potencial de acción cardiaco23. Alteraciones de estas proteínas de canal inducen cambios en la funcionalidad de los canales cardiacos, con ganancia o disminución de la función2428.
La célula cardiaca se despolariza a causa de una masiva y muy rápida entrada de cargas eléctricas positivas, principalmente a través del canal de sodio (Na+). Esto produce la fase 0 del potencial de acción. Inmediatamente se inicia la fase de repolarización, en la que la célula elimina cargas positivas porque tiene que volver al potencial de reposo. Éste es un proceso más lento y se realiza principalmente a través de un equilibrio entre los canales de potasio (K+) y calcio (Ca2+), lo que da lugar a las fases 1, 2 y 3 del potencial de acción29.
Son varios los elementos necesarios para conseguir una actividad cardiaca coordinada. Entre ellos, las corrientes iónicas, los canales iónicos y las proteínas estructurales encargadas de la transmisión del impulso eléctrico y mecánico a través de los miocitos cardiacos23. La complejidad de este proceso continúa siendo la mayor limitación para la comprensión de la arritmogénesis30.
Con la incorporación de la biología molecular a la cardiología, ya podemos resolver algunos de los misterios sobre la estructura, la función y la fisiopatología de los canales iónicos, lo que permite entender el papel que desempeñan éstos en la generación y la transmisión de la corriente eléctrica.
El análisis funcional de los canales iónicos ha permitido comprender mejor los mecanismos básicos arritmogénicos, pero no ha sido hasta el desarrollo de la genética y el descubrimiento de las mutaciones causantes de las enfermedades familiares cuando se ha podido extrapolar parte de la ciencia básica a la práctica clínica (tabla 1).

Las canalopatías, característicamente, no se acompañan de alteraciones cardiacas estructurales y su primera manifestación puede ser la muerte súbita. Además, algunas de estas enfermedades no se acompañan de alteraciones en el electrocardiograma (ECG) basal, lo cual dificulta aún más el diagnóstico. Teniendo en cuenta que estas enfermedades están determinadas por un defecto genético, es de esperar que las pruebas genéticas puedan contribuir sustancialmente al diagnóstico, la prevención y el tratamiento. Sin embargo, no se ha logrado la incorporación exitosa de estas pruebas a la práctica clínica para la evaluación de estas enfermedades.
Las arritmias cardiacas que predisponen a la muerte súbita se han beneficiado de los avances en genética y biología molecular31. Estas afecciones, generalmente monogénicas, han permitido estudiar la forma pura de la enfermedad, en la cual un solo gen causa la arritmia32, como el síndrome de Brugada, el síndrome del QT largo y el síndrome de QT corto (figs. 35) y la taquicardia ventricular polimórfica (TVP)24,25,31. Sin embargo, con la genética el conocimiento no se limita a la forma familiar, ya que abre nuevas hipótesis de cómo el gen interacciona con el ambiente, las medicaciones o el músculo dañado y causa la arritmia en las formas adquiridas o no familiares.

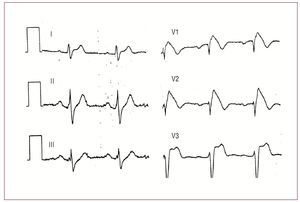
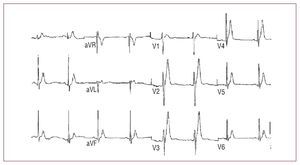
Fig. 3. Electrocardiogramas del síndrome de Brugada.

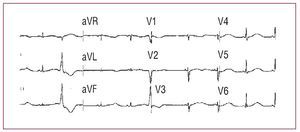
Fig. 4. Electrocardiograma del síndrome del QT largo.

Fig. 5. Electrocardiograma del síndrome del QT corto.
En general, en función del canal iónico afecto, podremos hablar de distintas afecciones. A pesar de esto, hay cierto solapamiento de un mismo síndrome donde diferentes tipos de canal pueden estar afectados.
Canalopatías por alteración de los canales de sodio
Los canales de sodio participan de una forma clave en la propagación del potencial de acción cardiaco. La apertura de estos canales permite la sincronización de la contracción ventricular y la velocidad de la conducción3336. Se han descrito varios tipos de mutaciones, con ganancia o pérdida de función del canal, las cuales dan lugar a arritmias37. Ciertas mutaciones pueden dar lugar a diferentes fenotipos, incluso combinaciones de ellos38.
Entre otras, tres enfermedades diferentes se han relacionado con una disfunción de los canales de sodio debido a las modificaciones genéticas en la estructura del canal: síndrome de QT largo, síndrome de Brugada y síndrome de LevLenègre.
Síndrome de QT largo
El síndrome de QT largo es una de las principales causas de muerte súbita entre los jóvenes. Puede ser congénito o adquirido, generalmente asociado a fármacos y desequilibrio hidroelectrolítico (hipopotasemia, hipocalcemia y hipomagnesemia)39. La presentación clínica puede ser variada, desde pacientes asintomáticos (diagnosticados en el contexto de un cribado familiar) a cuadros con síncopes, convulsiones, arritmias ventriculares malignas, fibrilación ventricular y, típicamente, torsades de pointes4045.
La forma congénita se asocia con mutaciones en los canales iónicos o proteínas relacionadas. La prolongación del inte rvalo QT puede surgir por una disminución en las corrientes repolarizadoras de potasio o por una inapropiada demora de la entrada de sodio en el miocito46.
Hasta la fecha se han descrito más de 500 mutaciones y 130 polimorfismos en el síndrome de QT largo dando lugar a 10 tipos distintos43. Aunque la mayoría de los trastornos que inducen QT largo se refieren a alteraciones en el canal de potasio, algunos tipos se asocian a alteraciones en los canales de sodio47.
El síndrome QT largo tipo 3 está asociado a mutaciones en el gen SCN5A48,49. La mutación causa un defecto funcional basado en una falta de inactivación completa del canal que permite que continúe la entrada de iones de sodio al interior celular durante la repolarización, con lo que se induce una ganancia de función50. Los pacientes con síndrome de QT largo tipo 3 presentan arritmias dependientes de bradicardia y síntomas en reposo (especialmente en la noche)51.
El QTL10 está causado por una mutación en SCN4B que codifica la subunidad beta (NaVβ4) del canal de sodio. La subunidad beta tiene un papel importante en la regulación de la cinética de canal, la transducción de señales y la expresión de la subunidad a del canal de sodio. La subunidad NaVβ4 provoca un cambio negativo en el voltaje de la dependencia de sodio en el canal de activación. Esta mutación en SCN4B induce un cambio positivo en la inactivación de los canales de sodio, lo que aumenta el INa y retrasa la repolarización de manera similar a lo que ocurre en el síndrome de QT largo tipo 352.
El síndrome QTL9 se produce por alteración de caveolina-3; se cree que este tipo aumenta el intervalo QT, ya que afecta a la funcionalidad de los canales de sodio. Las mutaciones en este gen conducen a una ganancia de función de los canales de sodio53, similar a lo que ocurre en el QT largo tipo 3.
Síndrome de Brugada
El síndrome de Brugada, descrito en 199254, se caracteriza por los hallazgos electrocardiográficos (elevación del segmento ST en V13) sin cardiopatía estructural. Es una importante causa de muerte súbita, generalmente por TVP, con una incidencia de alrededor de 2638/100.000 personas/año. Aunque la media de edad al inicio de los eventos es alrededor de los 40 años, la muerte súbita puede afectar a personas de todas las edades, especialmente varones (75%). De los pacientes afectados, un 2050% tiene antecedentes de muerte súbita familiar.
El síndrome de Brugada es una enfermedad de herencia familiar autosómica dominante y penetrancia variable55,56. Se han descrito más de 70 mutaciones distribuidas en varios genes, lo que demuestra la heterogeneidad genética57. Pese a que la mayoría de las mutaciones se producen en genes relacionados con los canales de sodio, el síndrome de Brugada también puede producirse porque se afectan otros canales. Esto demuestra que la fisiopatología de esta entidad posiblemente sea multifactorial por interacción entre varios mecanismos.
Un 2025% de los pacientes afectados por el síndrome de Brugada presenta mutaciones en el gen SCN5A58. Las mutaciones conllevan un cierre prematuro o que no se active el canal, lo que da una pérdida de función de los canales de sodio55,58. Esto induce un acortamiento de la fase 1 del potencial de acción, lo que deja la corriente de potasio Ito sin oposición en esta fase59 y resulta en la creación de un gradiente de voltaje, que es el sustrato ideal para generar arritmias por reentrada49. Además de las mutaciones también se han descrito varios polimorfismos que alteran la función del canal de sodio60 y podrían explicar los diferentes fenotipos clínicos e incidencia del síndrome de Brugada en diferentes zonas geográficas61, como en el sudeste asiático, donde se ha descrito una incidencia muy alta de muerte súbita por síndrome de Brugada62.
Otro gen relacionado con el canal de sodio y que ha sido descrito como inductor del síndrome de Brugada es GPD1-L. Se ha demostrado que la mutación de GPD1-L reduce la superficie de membrana de expresión y reduce el perfeccionamiento activo de sodio63. Además, GPD1-L se ha visto que es origen de una parte de las muerte súbitas del lactante64.
Síndrome de Lev-Lenègre
El síndrome de LevLenègre es una rara entidad que se caracteriza por una alteración del sistema de conducción que lleva al bloqueo gradual de éste, arritmias ventriculares o asistolia48,65,66. La
cantidad y la rapidez con que el sodio entra en la célula determinan la velocidad de conducción del impulso eléctrico a través de las células dependientes de sodio (células musculares del ventrículo y la aurícula y células del sistema HisPurkinje). Si una mutación produce una reducción en la cantidad de sodio que entra en la célula, se produce una disminución en la velocidad de conducción del impulso que da como resultado la pérdida de función en la fase 0 del potencial de acción (apertura del canal)67. Ése es el caso del síndrome de LevLenègre.
En 1995 se describieron por primera vez alteraciones cromosómicas (19q13.213.3) que llevan a bloqueo de rama68, y en 1999, las primeras mutaciones, localizadas en el gen SCN5A58.
Canalopatías por alteración de los canales depotasio
Los canales de potasio participan de una forma clave en el potencial de acción cardiaco, ya que permiten que las corrientes de repolarización contrarresten la despolarización anterior6973. Las mutaciones en los genes que codifican para las proteínas del canal de potasio pueden llevar a una disfunción de este canal y resultar en tres tipos de enfermedades: síndrome de QT largo, síndrome de QT corto y fibrilación auricular (FA).
Síndrome de QT largo
El síndrome de QT largo, la mayoría de veces, lo inducen alteraciones en la repolarización que implican a los canales de potasio (Iks, Ikr, Iki)45.
Todas las mutaciones en estos canales producen una pérdida de función; esto origina un descenso de la liberación de potasio desde las células que induce que los canales se mantengan abiertos más tiempo y se observe una prolongación del intervalo QT debido a un tiempo de repolarización ventricular más prolongado41,42.
Se han descrito multitud de mutaciones; de éstas, unas 300 se localizan en 6 genes de potasio diferentes que suponen un 5060% de los casos clínicos de QT largo7477.
Uno de ellos es el gen KCNQ1 (KvLQT1), el cual se une a la proteína codificada por el gen KCNE1 (minK) para formar el complejo funcional Iks. Las mutaciones en KCNQ1 producen un 4050% de los casos de QT largo, dando el síndrome QT largo tipo 178, el más común de los tipos de QT largo79, caracterizado por una repolarización retrasada y el consecuente QT prolongado80. Cuando su herencia es autosómica dominante, hablamos del síndrome de RomanoWard; cuando es autosómico recesivo da el síndrome de Jervell y LangeNielsen, típicamente asociado a sordera81.
Recientemente se han descrito seis nuevas mutaciones, dos exónicas y cuatro intrónicas82. En KCNE1 se han descrito hasta ahora cinco mutaciones83, las cuales inducen un 25% de los casos del llamado síndrome QT largo5, del que se cree que puede alterar tanto Iks como Ikr84.
Otro gen afectado es KCNH2 (HERG, human-ether-a-go-go-related)85, que codifica la subunidad alfa del complejo Ikr; la subunidad alfa está determinada por el gen KCNE2 (MiRP1)86. Este complejo Ikr es el mayor inductor de la repolarización rápida de la fase 387. Las mutaciones en KCNH2 (más de 80) dan una pérdida de función del canal Ikr, y suponen un 3545% del denominado síndrome de QT largo 2 de herencia autosómica dominante88. En KCNE2, la mutación induce también pérdida de función del canal, que da lugar al síndrome QT largo 6, un síndrome muy poco común (< 1%)79.
Otro gen implicado en el síndrome de QT largo es KCNJ2, situado en el cromosoma 17; codifica por Ik1 (Kir2.1), y contribuye en la fase 3 de repolarización, manteniendo el potencial de membrana. Las mutaciones en este gen suponen pérdidas de función que dan lugar al síndrome de QT largo 7 o síndrome de TawilAnderson89. Tiene muy baja incidencia en la población y raramente se relaciona con síncopes o muerte súbita90, aunque sí con episodios de taquicardias polimórfica o bidireccional91.
Síndrome de QT corto
Descrito en 200092, el síndrome de QT corto es una entidad de alta malignidad, caracterizada por un intervalo QT corto (< 330 ms), con onda T alta y picuda y el intervalo entre el pico y el final de la onda T no prolongado, provocando arritmias ventriculares y muerte súbita43,93.
La mayoría de los pacientes con síndrome de QT corto tienen una historia familiar de muerte súbita y/o FA. La edad a la aparición de manifestaciones clínicas puede ser la infancia, por lo que se ha catalogado como una posible causa de muerte súbita del lactante. La gravedad de las manifestaciones clínicas del síndrome de QT corto es muy variable, desde asintomático a la FA, síncope recurrente y muerte súbita.
El origen genético de esta entidad ha sido descrito recientemente, con patrón de herencia autosómico dominante y alta penetrancia94,95. Las mutaciones que inducen este síndrome se localizan en cinco genes43, de los cuales tres (KCNQ1,KCNJ2 y KCNH2) codifican para canales de potasio, con ganancia de función y, por lo tanto, acortamiento de la repolarización96,97.
El síndrome de QT corto tipo 1 se ha relacionado con mutaciones en el gen KCNH2(HERG) que inducen una rápida activación de las corrientes de potasio, con ganancia de función de IKr y un acortamiento de los potenciales de acción ventriculares98100. Generalmente, los eventos cardiacos se asocian a situaciones adrenérgicas como el ruido o el ejercicio, aunque también se han descrito en reposo101. El síndrome de QT corto se ha asociado a FA en algunas familias.
El síndrome de QT corto tipo 2 se ha relacionado con dos mutaciones en el gen KCNQ194,99,100 que comportan una ganancia de función del canal de potasio, lo que lleva a un acortamiento del potencial de acción con FA. Existe una entidad particular por alteración de este mismo gen que se manifiesta in utero en forma de bradicardia, que en el periodo neonatal se diagnostica de FA y QT corto99,100.
El síndrome de QT corto tipo 3 se ha relacionado con mutaciones en el gen KCNJ2 localizado en el cromosoma 17, comportando una aceleración de la fase 3 del potencial de acción102.
Fibrilación auricular
La FA es la arritmia más común observada en la práctica clínica. La prevalencia general del 1% aumenta con la edad hasta un 10% en la población de más de 80 años. La complicación más temida es la embolia cerebral y se considera que un tercio de todas las embolias se deben a ella103.
En 1997 se la describió por primera vez como una enfermedad genética con un patrón de herencia autosómico dominante104,105. A pesar de esto, son numerosos los genes que se han relacionado con la FA, sobre todo los que codifican para los canales de potasio (KCNQ1, KCNE2, KCNJ2 y KCNH2)106. Asimismo, se ha visto que los factores ambientales son especialmente modificadores de la presentación y la evolución107109. Además, se ha descrito que las mutaciones o los polimorfismos en el gen SCN5A pueden predisponer a los pacientes a la FA110, aunque las variaciones en el gen SCN5A no parecen ser una causa importante de FA familiar17,18.
Canalopatías por alteración de los canales de calcio
Los canales de calcio se han visto relacionados con un número cada vez mayor de arritmias cardiacas familiares111.
Los iones de calcio participan en la fase 2 del potencial de acción cardiaco e incrementan la salida de calcio del retículo sarcoplásmico que permite iniciar la activación del aparato contráctil del corazón. Con unas funciones tan relacionadas con la actividad electromecánica, era de esperar que las alteraciones en las proteínas que intervienen en el equilibrio de calcio pudieran originar las arritmias cardiacas112.
Se han visto relacionados en los canales de calcio: combinación síndrome de Brugada y QT corto; síndrome de Timothy, y TVP.
Combinación síndrome de Brugada y QT corto
La mutación en el gen CACNA1C causa una alteración en la unidad alfa de los canales de calcio tipoL induciendo una pérdida de función del canal, relacionada con la asociación al síndrome de Brugada y QT corto tipo 4, con patrón de herencia autosómica dominante. Con el mismo fenotipo, la mutación en CACNB2b causa una alteración en la unidad de los canales de calcio tipoL, y se produce la combinación síndrome de Brugada y QT corto tipo 5111,113,114.
Síndrome de Timothy
El síndrome de QT largo 8 o síndrome de Timothy es una forma de QT largo de descripción reciente115, en que las alteraciones se deben a una mutación en el gen CACNA1C que codifica para el poro (Cav1.2) del canal de calcio cardiaco tipoL116.
Este tipo de QT largo es poco frecuente, pero es el más letal. La mutación induce una ganancia de función con alteración de Ica, con pérdida dependiente de voltaje del canal que origina una prolongación del potencial de acción que da lugar a un ECG con un QT largo severo.
Taquicardia ventricular polimórfica
La taquicardia polimórfica catecolaminérgica es un trastorno arritmogénico familiar caracterizado por una taquicardia ventricular bidireccional y polimórfica117, desencadenada exclusivamente por el estímulo adrenérgico (ejercicio vigoroso, miedo), con alta mortalidad (el 30% a los 30 años de edad).
Se han identificado dos variantes genéticas, una autosómica dominante causada por mutación en el gen del receptor de la rianodina RyR2 (1q42Q43) y una recesiva,or mutación en la isoforma del gen de la calciquestrina (CASQ2)118121. Ambos genes están implicados en la regulación del calcio intracelular y ambos defectos generan un aumento en la función de estas proteínas, por lo que se incrementa la salida de calcio del retículo sarcoplásmico. Este exceso de calcio se relaciona con alteraciones en el potencial de membrana del sarcolema, con aparición de despolarizaciones tardías que facilitan las arritmias120.
El receptor de la rianodina es un canal de calcio intracelular que se encuentra en el retículo sarcoplásmico y se activa por la entrada de pequeñas cantidades de calcio permitiendo la salida del calcio almacenado122. Se han identificado más de 70 mutaciones en RyR2. En el corazón, el receptor de la rianodina se asocia a dos enfermedades diferentes: la displasia arritmogénica de ventrículo derecho (DAVD) tipo 2 (DAVD2)123 y la TVP familiar119. Es interesante que el mismo gen cause dos enfermedades tan distintas, una con alteración estructural —la DAVD2— y la otra sobre corazón estructuralmente normal. En la actualidad se está investigando si esta diferencia se debe al tipo de mutación, a modificadores genéticos o al ambiente.
Otros genes que inducen canalopatías
Existen otros genes como el ANK2 (cromosoma 4, 4q2527), que está involucrado en el síndrome de QT largo 4. A pesar de no afectar específicamente a un canal, se incluye en el grupo de las canalopatías. Este gen codifica la proteína ankirina B, cuya función es adaptar distintas estructuras en la membrana c elular, como la bomba Na/K ATPasa, intercambiador Na/Ca y receptor del inositol trifosfato124,125. Una disminución en la función de la ankirina B altera la homeostasis de calcio, prolonga la repolarización y genera arritmias ventriculares letales126.
Otro gen es Caveolina-3 (Cav3), implicado en el tráfico de membrana y con la correcta posición de las proteínas de los canales iónicos situados en la membrana sarcoplásmica. Las mutaciones inducen una ganancia de función de los canales de sodio, dando el síndrome QT largo tipo 953.
Miocardiopatías
Se han detectado varias mutaciones que causan miocardiopatías arritmogénicas en el ser humano127; estas mutaciones se han identificado en un amplio número de genes que codifican tanto para proteínas contráctiles y estructurales como para la producción de energía cardiaca (fig. 3; tabla 2).

Miocardiopatía hipertrófica
La MCH es una enfermedad del miocardio caracterizada por una inexplicada hipertrofia asimétrica del ventrículo izquierdo, con hallazgos de desestructuración de los miocitos y fibrosis128,129. Es la alteración genética cardiovascular más frecuente, con una prevalencia en la población general es de 1/500130134, afectando a niños y jóvenes. Las manifestaciones clínicas aparecen por disfunción diastólica inicialmente y sistólicodiastólica en estados más evolucionados, por lo que el paciente puede estar asintomático o presentarse en forma de insuficiencia cardiaca o muerte súbita. La mortalidad es mayor en los pacientes jóvenes (a menudo atletas) que en los adultos, y la primera manifestación de la enfermedad puede ser precisamente la muerte súbita. La enfermedad se considera familiar en un 90% de los casos, generalmente con un patrón de herencia autosómica dominante, a excepción de los casos con mutaciones en el ADN mitocondrial (ADNmt), que se heredan por vía materna.
La entidad clínica fue descrita en 1958135, pero no fue hasta 1989 cuando se descubrió el primer gen implicado en esta enfermedad. Desde entonces se han identificado más de 400 mutaciones136. A pesar de esto, el 60% de los pacientes con MCH presentan alteraciones en sólo 9 genes, en los que se centrará el cribado genético.
Estos genes codifican para las proteínas de la estructura sarcomérica del músculo cardiaco, como la cadena pesada betamiosina (MYH7)137,138 y la proteína C de unión a miosina (MYBPC3)133; otros codifican para la cadena pesada alfamiosina (MYH6), troponina I (TNNI3)139, troponina T (TNNT2)140,141, alfatropomiosina (TPM1)140,141, cadenas ligeras de miosina esencial (MYL3)142, regulatoria (MYL2), titina y alfaactina (ACTC)143146. También se han detectado mutaciones en genes involucrados en el metabolismo del grupo hemo y Fe2+, y en genes de la bioenergética mitocondrial.
Estudios genéticos en familias con hipertrofia ventricular izquierda han demostrado cardiomiopatías metabólicas con mutaciones en PRKAG2 y en LAMP2. Hasta ahora se considera que las mutaciones no predicen el fenotipo, ya que en una misma familia pueden existir individuos con diferentes grados de hipertrofia o con mayor predisposición a muerte súbita que otros con la misma mutación. Esto se debe a la intervención de genes modificadores y polimorfismos, que requieren estudios más exhaustivos128.
Con base en estos resultados, se asume que la interrupción del metabolismo energético mitocondrial cardiaco es la causa de MCH en los pacientes con problemas de contracción sarcomérica; esto nos puede ayudar a entender numerosas observaciones clínicas como su heterogeneidad, su variabilidad en la presentación clínica y su asimetría en la hipertrofia. La identificación del genotipo podría contribuir a la estratificación de riesgo, pero deben realizarse más estudios de genotipofenotipo para confirmar esta utilidad.
Miocardiopatía dilatada
La MCD está caracterizada por dilatación ventricular que comporta una alteración de la función sistólica, principalmente en el ventrículo izquierdo. Los pacientes presentan signos de insuficiencia cardiaca, palpitaciones o muerte súbita. La prevalencia es de 1/2.500 personas. Son múltiples los factores que pueden desencadenas una MCD, por lo que es una entidad altamente heterogénea. A pesar de esto, los estudios sistemáticos de los familiares de pacientes con MCD indican que al menos un 35% de los casos son hereditarios147. Las arritmias que presentan los pacientes con MCD familiar suelen ser las mismas que en las formas adquiridas, con defectos en la conducción auriculoventricular e intraventricular, arritmias ventriculares y FA. En estos pacientes normalmente se produce un deterioro progresivo de la función ventricular y fallecen debido a insuficiencia cardiaca o arritmias. La mortalidad a los 5 años es de entre un 40% y un 80%.
En 1994 se encontró el primer locus de MCD con bloqueo auriculoventricular en el cromosoma 1148. El escenario de esta enfermedad es sumamente complejo, por lo que la utilidad clínica del análisis genético es limitada. Se han identificado más de 20 mutaciones en genes que codifican para proteínas del citoesqueleto, núcleo celular y sarcómero. Una de las mutaciones más importantes (30%) es la encontrada en el gen lamina A/C (LMNA)148,149, que codifica una proteína que se expresa en casi todos los tipos celulares y cuya función es contribuir a la integridad del núcleo proporcionándole soporte mecánico150,151. Otros genes, como MYH7 y TNNT2, identificados previamente como causa de MCH, también pueden causar MCD. Incluso en una gran familia con MCD se identificó mutación en SCN5A152.
Los patrones de herencia de las formas familiares son varios: a) enfermedad autosómica dominante, de la que se han descrito locus en varios cromosomas (actina, desmina, lamina A/C, deltasarcoglucano); b) enfermedad ligada al cromosoma X, en la que se ha descrito el gen de la distrofina como causante de la enfermedad. Mutaciones directas en el gen de la distrofina originan distrofia muscular de Duchenne o de tipo Becker, en las cuales se afectan tanto el músculo cardiaco como el esquelético; sin embargo, las mutaciones en este gen no son una causa común de MCD, y c) enfermedades mitocondriales, las cuales típicamente afectan a otros órganos además del miocardio; hasta ahora se han descrito dos loci de MCD conjuntamente con arritmias primarias, sin que una sea la causa de la otra. Estas familias presentan la enfermedad de forma autosómica dominante.
Displasia arritmogénica de ventrículo derecho
La displasia arritmogénica del ventrículo derecho (DAVD) es una enfermedad que se caracteriza por sustitución progresiva del tejido miocárdico por tejido adiposo y fibrosis, con afección progresiva del epicardio hacia el endocardio, típicamente en el ventrículo derecho21,153,154. Suele afectar a 1/5.000 personas, aunque con una prevalencia aumentada en varones (80%). La mayoría de los casos son diagnosticados antes de los 40 años de edad. Normalmente los individuos afectados presentan arritmias ventriculares sintomáticas que se inician en el ventrículo derecho, síncope y gran riesgo de muerte súbita155,156. Es la causa del 5% del total de muertes súbitas, especialmente en jóvenes atletas.
La enfermedad tiene dos patrones de herencia distintos. La forma más común es la autosómica dominante, de la que se han identificado hasta el momento mutaciones en seis genes, incluidos cuatro que codifican proteínas del desmosoma (proteínas de unión intercelular)157. En el 45% de los pacientes con esta enfermedad se encuentra una mutación que afecta a la proteína plakofilina 2 (PKP2)158. El resto de los defectos genéticos se identifican en una proporción muy baja de pacientes y se deben a defectos en proteínas como desmoplaquina, placoglobina, desmogleína2 o desmocollina2159.
Al haber tantos loci involucrados en la enfermedad, se ha descrito varios fenotipos de DAVD: el DAVD tipo 1 por alteración en 14q23-24160, el tipo 2 por alteración en 1q42-q43123, la DAVD tipo 3 por mutación en 14q12-q22161, el tipo 4 por alteración en 2q32162, el tipo 5 por mutación en 3q23163, el tipo 6 que afecta a 10p12-p14164, la DAVD tipo 7 por mutación en 10q22.3165, el tipo 8 con alteración en 6p24166, el tipo 9 por alteración en 2p11167, el tipo 10, por mutación en 8q12.1q12.2168 y, finalmente, la DAVD tipo 11 por mutación en 18q21169.
Se ha descrito una forma con patrón de herencia autosómica recesiva en la isla griega de Naxos, denominado precisamente síndrome de Naxos. Este síndrome consta de DAVD, queratoderma palmoplantar y un cabello rizado típico.
El análisis genético puede ser útil para el cribado familiar, ya que la incidencia familiar es del 50%, lo cual permite establecer el diagnóstico en los casos indeterminados y en portadores de la mutación asintomáticos. En estos individuos es importante el consejo genético previendo futuros embarazos. Son necesarios estudios de correlación genotipofenotipo para valorar el papel del análisis genético en esta enfermedad.
CONCLUSIONES
El avance en el conocimiento del genoma humano ha permitido abrir numerosas vías de investigación genética para todo tipo de enfermedades. El campo de la cardiología es uno de los que más se ha beneficiado del tremendo potencial de la genética y de la aplicación de la tecnología genéticomolecular.
Ha pasado ya más de una década desde el descubrimiento del primer gen que causa una enfermedad familiar cardiaca y el nuevo enfoque multidisciplinario en el manejo de estas enfermedades comprende la integración de la investigación tanto básica como clínica, abriendo nuevas posibilidades para la prevención, la estratificación de riesgo, el diagnóstico y el tratamiento170. Entender las enfermedades cardiovasculares a escala genómica puede permitirnos hacer una mejor estratificación de subclases de pacientes para optimizar y dirigir terapias específicas para cada paciente20,171.
Se espera que futuras investigaciones generen un cambio en la forma en que luchamos contra estas enfermedades. Los campos relacionados con la farmacogenética abrazan la promesa de mejorar el desarrollo de fármacos personalizados que tengan en cuenta la edad y la genética individual que modifican el transporte y metabolismo de los medicamentos. La meta no es eliminar todos los genes causantes de enfermedad, sino permitir la transición desde un cuidado paliativo y reactivo hacia los tratamientos preventivos que disminuyan o eviten la expresión de mutaciones o polimorfismos relacionados con mayor predisposición a las arritmias letales172.
Son tres los factores clave en el avance de la biomedicina hacia los tratamientos personalizados: pacientes, clínicos e investigadores básicos. La interacción entre ellos permitirá la mejora del tratamiento de las diferentes enfermedades.
Full English text available from: www.revespcardiol.org
Sección patrocinada por el Laboratorio Dr. Esteve
Correspondencia: Dr. J. Brugada Terradellas. Servicio de Cardiología. Hospital Clínico y Provincial c/ Villarroel, 170. 08036 Barcelona. España.
Correo electrónico: jbrugada@clinic.ub.es