Chronic heart failure (CHF) hospitalizations continue to grow in Europe and the United States. It is estimated that hospitalizations for acute decompensated heart failure (ADHF) account for >75% of the health care costs for CHF patients.1,2 Data from large trials and registries have shown that most hospitalizations for ADHF occur because of symptoms (dyspnoea, abdominal bloating, and fatigue) and signs (pulmonary rales, jugular vein distension, and peripheral edema) of venous congestion rather than of low cardiac output.3,4 Symptoms of congestion typically worsen a few days (3 [2.5] days) before hospital admission.5 However, recent studies have shown that the onset of venous congestion occurs well before symptoms of congestion become apparent. Home monitoring of daily weight,6 and continuous monitoring of intracardiac pressures (Chronicle, Medtronic Inc.),7 and pulmonary congestion via intrathoracic impedance, (OptiVol, Medtronic Inc.),5 all provide evidence that venous congestion begins to occur much earlier than previously thought in the time course of ADHF.
Venous congestion (marked by an increase in weight, right side filling pressures, and accumulation of intrathoracic fluid) starts to increase at least 7- 14 days before CHF signs and symptoms worsen, eventually requiring urgent intravenous therapy.5-7 Although congestion is an important target of treatment, physicians do not do a very good job in treating congestion, as evidenced by the fact that ≈50% of patients do not lose body weight during hospitalization.8 This treatment failure has major consequences, as refractory systemic congestion is an important hemodynamic predictor of worsening renal function, rehospitalization and post-discharge mortality in patients hospitalized for ADHF.9-13
Dietary indiscretion, medication noncompliance, ischemia, arrhythmias, and worsening hypertension and left and/or right ventricular systolic or diastolic function may all promote fluid retention and venous congestion in patients with CHF.14 However, while fluid accumulation represents the effect rather than the cause, once initiated and sustained, it exercises negative effects on the heart (eg, by promoting subendocardial ischemia),15 on the kidneys (eg, by reducing perfusion pressure and causing sodium retention),12,13,16 and also, based on our initial observations, on the venous endothelium and on the peripheral production and release of cytokines and neurohormones.17,18
Our hypothesis is that venous congestion itself is a fundamental inflammatory and hemodynamic stimulus that contributes to the development and progression of ADHF through endothelial, neurohormonal, renal, and cardiac mechanisms. The discussion that follows will detail the evidence supporting this hypothesis.
The venous endothelium is the largest endocrine/ paracrine organ of the body and a key regulator of central blood volume, organ perfusion and haemostasis in CHF through transitions between quiescent and activated states that occur in response to environmental stressors such as vascular stretch associated with venous congestion.19 Using a novel approach that involves sampling of venous endothelial cells and quantification of protein and mRNA expression, we have previously reported an increase in pro-oxidant free radicals and pro-inflammatory proteins in venous endothelial cells collected from patients hospitalized for ADHF with clinical evidence of fluid overload.20,21 This venous endothelial activation partially subsides with diuresis and clinical improvement during the index hospitalisation.21 More recent animal and human data confirmed that venous congestion itself can switch the synthetic and endocrine profile of the endothelium, from quiescent toward an activated state that is pro-oxidant, pro-inflammatory and vasoconstricting in a manner consistent with that seen in patients with ADHF.17,18 The resulting peripheral release of vasoactive and pro-inflammatory neurohormones (ie, tissue necrosis factor-a, endothelin-1, interleukin-6, and angiotensin II), from the stretched endothelium and from the perivascular congested tissue, may offset the physiological adaptations that sustain the compensated state of CHF (ie, redistribution of limited cardiac output to vital organs such as the kidneys, heart, and brain). These pathophysiological events may promote additional fluid retention, and leading to a vicious cycle that can eventually result in overt decompensation.
From a renal standpoint, an increase in venous congestion and pressure has both hemodynamic and intrinsic detrimental consequences on the kidneys as it reduces organ perfusion and increases Na retention.16 Of note, both pro-oxidant free radicals and pro-inflammatory cytokines such as tissue necrosis factor-a, which are released by the stretch endothelium in response to venous congestion, have been shown to reduce renal sodium excretion.22,23
Venous congestion may also negatively impact cardiac function by causing subendocardial ischemia, left ventricular remodelling, impairment of cardiac venous drainage from coronary veins, and a lower threshold for arrhythmias.15 The resulting decrease in cardiac output may further impair renal perfusion and function, thereby causing additional fluid retention. Of note, the cardiac endocardium is structurally identical to and in continuity with the vascular endothelium, and is likely to be activated and possibly contributes to the release of angiotensin II that occurs in response to elevated cardiac filling pressures.24
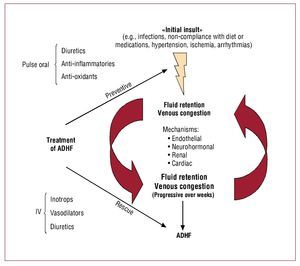
Figure 1 summarizes what we consider as a "unifying peripheral venocentric hypothesis" for the pathophysiology of ADHF. At the beginning there is an insult, that causes fluid retention maybe multiple ones, of varying etiology (eg, infections, non-compliance with diet or medications, arrhythmias, ischemia, and/or worsening hypertension). Through endothelial, neurohormonal, renal, and cardiac mechanisms, venous congestion itself may lead to additional sodium and water retention. Vascular stretch associated with venous congestion may switch the synthetic and endocrine profile of the venous endothelium, from quiescent toward an activated pro-oxidant, pro-inflammatory and vasoconstricting state that, in turn, promotes peripheral release of vasoactive and pro-inflammatory neurohormones. In the kidneys, vascular congestion and activation of the stretched endothelium, now itself a source of oxidative stress and pro-inflammatory cytokines, may cause additional fluid retention. In the heart, high filling pressure further impairs systolic and diastolic function. Subsequently, when the initial insult(s) subside(s), it may be too late: the vicious cycles that link venous congestion to progressive fluid retention are already in place. Symptoms will eventually worsen after weeks of progressive fluid accumulation, eventually leading to hospitalization for overt decompensation.

Figure 1. Impact of venous congestion on the pathophysiology and treatment of acute decompensated heart failure (ADHF).
Early detection (eg, by continuous monitoring of intracardiac pressures and intrathoracic impedance) and better understanding of the pathophysiology of ADHF may allow future treatment strategies to switch from the current "rescue mode" of late intravenous interventions (ie, inotropes, diuretics), to a "preventive mode" of oral interventions. This early treatment strategy may include not only diuretics but also, as one may infer from our data and test in future studies, adjuvant means such as short-term (pulse) anti-oxidant and/or anti-inflammatory treatments that may abort ADHF before the progression to overt decompensation.
In conclusion, venous congestion may act as an independent and fundamental stimulus for the development and progression of ADHF. Importantly, our "venocentric" approach to the pathophysiology of ADHF is aimed at complementing rather than replacing the more traditional "cardiocentric," "nephrocentric," "arteriocentric" views, since all systems (ie, the heart, the kidneys, the arteries, and the veins) appear involved in the events that trigger and sustain ADHF.
Disclosure: Paolo C. Colombo, MD: Medtronic Inc: Investigator-Initiated Research Grant.
Correspondence: Paolo C. Colombo, MD,
Division of Cardiology, New York-Presbyterian Hospital, Columbia University
622W 168th Street, PH 12-134, New York, NY 10032, USA
E-mail: pcc2001@columbia.edu