Keywords
Arteriosclerosis—a complex chronic disease of the vessel wall—is the underlying cause of many cardiovascular episodes, including coronary artery disease (Figure 1). The etiology of atherosclerosis is multifactorial and the trigger can be either systemic or local factors that induce deterioration in vascular function. Hypercholesterolemia and especially elevated plasma concentrations of low-density lipoproteins (LDL) constitute an important risk factor for the premature onset of atherosclerosis and ischemic heart disease.1

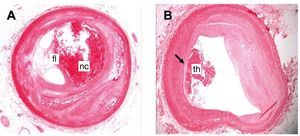
Figure 1. Human coronary arteries with different types of atherosclerotic lesion. A: vulnerable complex atherosclerotic plaque, with large necrotic center (nc) and a fine fibrous layer (fl). B: eroded plaque with superimposed thrombus (th). Hematoxylin-eosin staining.
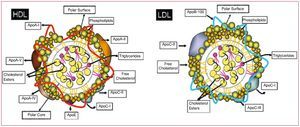
Lipids are insoluble organic molecules whose very low solubility in aqueous media results from their highly hydrophobic nature. Lipoproteins are structures composed of proteins and phospholipids that facilitate the transport of lipids in the bloodstream. Specifically, LDL is a heterogeneous class of lipoproteins with a density between 1.019 and 1.063 g/mL and a diameter of 20-25 nm. LDL particles consist of a hydrophobic core that contains triglycerides and cholesterol esters, surrounded by a hydrophilic surface of phospholipids, free cholesterol, and proteins, mainly apolipoprotein B-100, which acts as a ligand for cell membrane receptors (Figure 2).

Figure 2. Schematic of high density lipoproteins (HDL) and low-density lipoproteins (LDL). Main protein and lipid components. Apo indicates apolipoprotein.
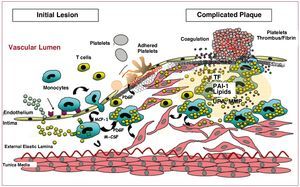
In regions of vascular wall predisposed towards atherosclerotic lesions, with increased permeability, hypercholesterolemia is associated with increased LDL transcytosis through the vascular endothelium. This leads to an accumulation of LDL particles in the subendothelial space, where they interact with proteoglycans and proteins that stimulate their modification (aggregation, glycosylation, enzymatic proteolysis, oxidation, etc), thereby increasing their atherogenicity2,3 and retention in the vascular intima. Retention of LDL in the arterial intima is a key process in initiating progression of the atherosclerotic lesion, as it triggers a local inflammatory process. Indeed, atherosclerotic lesions are the result of complex interactions between inflammatory cells, platelets, vascular elements, and lipoproteins that regulate the expression of genes and proteins directly involved in the vascular remodeling process (Figure 3).

Figure. 3. Schematic representation of the progression of atherosclerotic plaque from initial stages of endothelial dysfunction to advanced stages with the presence of complicated plaques. M-CSF indicates macrophage colony stimulating factor; MCP-1, monocyte chemotactic protein 1; MMP, metalloproteinases; PAI-1, plasminogen activator inhibitor 1; PDGF, platelet-derived growth factor; TF, tissue factor; UPA, urokinase plasminogen activator
LIPOPROTEINS AND ENDOTHELIAL DYSFUNCTION
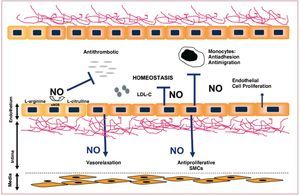
The vascular endothelium is a multifactorial organ able to perceive stimuli (both systemic and local) and modify its functional status to contribute to the homeostasis of the vascular wall.4,5 Under physiological conditions, the endothelium presents a surface with antithrombogenic properties.6 It allows exchange of numerous substances between the blood and tissues and controls the vascular tone and trafficking of inflammatory cells towards the vascular bed. The increased permeability of the endothelium in the presence of elevated concentrations of LDL has been recently associated with activity of kinase p21, through a mechanism mediated by protein kinase G and Ser/Thr kinase Akt.7,8 A large part of the antiatherogenic and antithrombotic properties of the vascular endothelium are mediated by its capacity to produce and release substances such as nitric oxide (NO), a platelet aggregation inhibitor with a strong vasodilatory activity and an important anti-inflammatory function. NO blocks the expression of proinflammatory molecules such as necrosis factor (NF) kB and adhesion molecules (ICAM-1, VCAM-1),9 as well as leukocyte adhesion and infiltration.10 Cell-cell connections such as tight junctions and gap junctions are essential structures in regulating the endothelial permeability function. The formation of gap junctions is regulated by the presence and functionality of connexins, proteins whose expression is altered during the formation of atherosclerotic lesions.11 Gap junctions, in addition to favoring intercellular signaling processes, regulate NO-dependent vasodilation (Figure 4).

Figure 4. Simplified diagram of the vasoprotective effects of nitric oxide. LDL-C indicates low-density lipoprotein cholesterol; NOS, nitric oxide synthetase.
Atherogenic concentrations of LDL lead to a decrease in the bioavailability of endothelial NO. This decreased NO availability is associated with a reduction in the concentration and/or activation of NO synthetase, as a result of the presence of native LDL13 or modified LDL14 particles, as well as the degradation of NO through the formation of superoxide anions (O2).15,17 The imbalance between NO/redox is associated with protein nitrosylation. Deactivation of NO -by O2 gives rise to highly cytotoxic peroxynitrite radicals. The presence of peroxynitrites derived from -nitrotyrosines and an increased production of O2 has been reported in human atherosclerotic lesions. In addition, peroxynitrites inhibit the expression of prostacyclin synthetase and favor its nitrosylation, a process that has recently been linked to superoxide-induced endothelial dysfunction in vascular disease.
RETENTION AND MODIFICATION OF LDL PARTICLES IN THE ARTERIAL INTIMA. EFFECT ON THE PROGRESSION OF ATHEROSCLEROTIC LESIONS
The vascular remodeling that takes place during the development and complication of atherosclerotic plaques is largely influenced by the composition and structure of the extracellular matrix (ECM). Smooth muscle cells (SMCs) produce most of the main components of the ECM, such as proteoglycans, collagen, and elastin, as well as a large number of proteins responsible for the equilibrium between synthesis (lysyl oxidase) and degradation (metalloproteinases, plasminogen activators) of the ECM during the atherogenic process.
LDL particles undergo modification processes when they interact with ECM components, because of the action of certain oxidants (lipoxygenase, myeloperoxidase, free radicals, etc), and/or because of proteolytic enzymes (kinase, tryptase, metalloproteinases, thrombin, etc), lipolytic enzymes (sphingomyelinase, phospholipase A2, phospholipase C, etc), and hydrolytic enzymes (esterase). Thus, certain chemical and/or structural processes take place that generate different types of modified LDL particles.18
Proteoglycans are responsible for trapping the LDL particles in the arterial intima. Chondroitin sulfate proteoglycans, such as versican, are the main structural proteoglycans of the ECM and are considered important atherogenic elements given that they can strongly interact with, retain, and aggregate cholesterol-rich lipoproteins; LDL particles of lowest density are those with the greatest capacity for interacting with the proteoglycans. The length and number of glycosaminoglycan chains in proteoglycans produced by SMCs, as well as their degree of sulfatation, will determine the formation of insoluble complexes between these molecules and the capacity for retention of the LDL particles in the arterial intima.19
The interaction between versican and LDL leads to structural changes in these lipoprotein particles and in turn to LDL aggregates similar to those obtained mechanically by shaking and to those described in atherosclerotic lesions.20
Collagen is essential for maintaining the integrity and elasticity of the vascular wall. However, it is also implicated in cell differentiation processes, and its glycosylated forms are an important factor in atherogenesis as they favor LDL retention.21,22
Unlike native LDL particles, which are internalized specifically by the LDL receptor, our group has shown that the forms modified by aggregation are internalized in cells by different types of receptors, including LRP-1 (low-density lipoprotein receptor related protein-1), an LDL family receptor that internalizes LDL aggregates,23 scavenger receptors such as SR
AI and SR-AII, CD36, LOX-1, or CXCL16 for oxidized LDL,24 and Fc gamma receptor for LDL incorporated into immune complexes.25 The LDLR, LRP-1, and scavenger receptors have been detected in different cell types such as monocytes, SMCs, and platelets, with key functions in the development of atherosclerotic lesions.
LDL AND MONOCYTES/MACROPHAGES
An important feature during the development of atherosclerotic lesions is the infiltration of circulating monocytes into the intravascular space. LDL particles, especially the modified forms, increase the expression and secretion of soluble chemotactic compounds (MCP-1, interleukin [IL] 8)26 and enhance the expression of adhesion molecules such as integrins and selectins,27,28 which are exposed on the surface of activated endothelial cells and favor leukocyte (monocyte and T-cell) recruitment, adhesion, and transmigration. The monocyte chemotactic protein (MCP) 1 plays a key role by means of the CCR2 receptors. The fact that different adhesion molecules and chemotactic molecules are expressed almost simultaneously in endothelial cells indicates a concerted activation of different genes through a common transcription factor such as NF-kB.29 In addition to the effects on the vascular endothelium, modified LDL particles directly favor the entry of monocytes in the vascular wall through a process that is thought to be mediated by CD11 and the protein kinase C (PKC) pathway.30
Diapedesis of monocytes takes place through the spaces (junctions) between endothelial cells, preferably in areas where the basal lamina is enriched with modified LDL particles.31 Then, infiltrated monocytes differentiate into macrophages and express scavenger receptors such as CD36 and LOX-1, which internalize many of the cholesterol molecules and cholesterol esters contained in modified LDL particles. LDL aggregates (LDLag) are potent inducers of massive cholesterol accumulation in macrophages32; whereas some authors propose phagocytosis as the classic mechanism of LDLag internalization in macrophages, other authors, using experimental designs that simulate the in vivo interaction between macrophages of the wall and subendothelial LDL particles, have defined a new internalization mechanism known as "pathocytosis," which consists of capturing large amounts of LDLag by means of vesicular membrane formations.33 Recently, it has been reported that LRP-1 is involved in macrophage internalization of LDL retained in the matrix and/or degraded by sphingomyelinase34 in a process regulated by SREBP1 and SREBP2, which in turn control LRP-1 expression.35 Cholesterol internalization leads to the formation of foam cells, a characteristic cell constituent of atherosclerotic lesions. In turn, foam cells secrete proinflammatory cytokines, growth factors, tissue factor,36 interferon (IFN) q, MMP, and reactive oxygen species (ROS) that maintain the chemotactic stimulus for leukocytes adhered to the vascular endothelium, increase the expression of scavenger receptors, enhance macrophage replication,26 and regulate SMC accumulation in the intima.37
LDL AND SMCs
SMCs are the major component of the vascular wall, and therefore have a key role in vascular function. SMCs are implicated in the initiation, progression, and complication of the atherosclerotic lesions. SMCs are characterized by their high plasticity. Under the effects of atherogenic stimuli, SMCs undergo phenotypic changes ranging from differentiation to the acquisition of a synthetic phenotype. Thus, SMCs with a nonproliferative contractile phenotype, typical in healthy arteries, transform into actively proliferative cells (synthetic phenotype), migrate attracted by chemotactic agents, and have increased ECM synthesis. In fact, migration of SMCs from the vascular media to the vascular intima is a key process in the development of atherosclerotic lesions. Recent data indicate that, in addition to migration of resident SMCs from the vascular media to the intima, circulating progenitor cells from bone marrow and progenitor cells present in the vessel adventitia are also a potential source of SMCs in the intima.38
Functional studies with genetically modified animals or cell culture studies demonstrate that certain components of the LDL-related receptors modulate cell migration by means of signaling processes mediated by cytokines and activation of proteinases.39
In the presence of LDLag, SMCs overexpress receptors such as LRP-1 which, in addition to facilitating LDL internalization and the subsequent transformation into foam cells,20 acts as a receptor to different ligands and participates in signaling processes. In hypercholesterolemic animals, messenger RNA analysis by in situ hybridization shows an increase in vascular wall LRP-1,20indicating that hypercholesterolemia might favor LDL capture by SMCs through its capacity to regulate the amount of LRP-1. In addition, greater expression of the LRP-1 receptor might be influencing other important processes in atherothrombosis, as LRP-1 mediates the internalization and degradation of relevant molecular complexes in proteolysis and fibrinolysis. Thus, LRP-1 can interact directly or indirectly with different members of the MMP family, and its participation in the catabolism of MMP9 in fibroblasts has been demonstrated.
SMCs and MMP9 in Atherogenesis
MMP9 is a metalloproteinase belonging to the gelatinase group, which is expressed by SMCs and macrophages in the vascular wall. It is latently secreted to the ECM, where it is activated by plasmin in a process mediated by the urokinase plasminogen activator. MMP9 is overexpressed in restenosis and SMC migration. Recent evidence shows that induction of the transcription of MMP9 by cytokines and atherogenic growth factors is regulated by the Foxo4 through the JNK/MST1 signaling pathway.40 MMP9 is a proteolytic enzyme of different ECM-related proteins such as type IV collagen, laminin, and elastin,41 which favor remodeling of the vascular wall. Thus, apoE-/- MMP9 knockout mice have smaller atherosclerotic lesions than apoE-/- mice with normal expression of MMP9.42
SMCs and the UPA/UPA Receptor (UPA/ UPAR) System in Atherogenesis
UPA, bound to its receptor UPAR (CD87), regulates mechanisms necessary for cell migration and proliferation either directly or in a process mediated by the generation of plasmin through pericellular proteolysis.43 In addition, there is increasing evidence that the UPA/UPAR system acts as a ligand-inducer of cell signaling mechanisms in proliferation and migration44 (Figure 5).

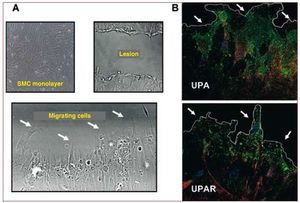
Figure 5. Localization of UPA and UPAR in SMCs of human coronary arteries in migration. A: phase contrast microscopy showing cells migrating to a lesion zone (lacking cells) induced mechanically in a confluent human SMC monolayer. B: confocal fluorescence microscopy for UPA and UPAR (green signal) and actin (red signal) in cells on the edge of the lesion (indicated by arrows). Nuclei stained blue with Hoescht blue fluorescent dye. SMC indicates smooth muscle cell; UPA, urokinase plasminogen activator; UPAR, urokinase plasminogen activator receptor.
A soluble form of the LDL receptor family (ie, LR11 or sorLA) highly expressed in SMCs, increases SMCs' capacity for migration in a process mediated by an increased UPAR expression. Studies on LR11 knockout mice indicate that the soluble forms of LR11 favor the formation of lamellipodia of the SMCs forming LR11-UPAR-integrin (anb3) complexes and that this process is necessary for SMCs adhesion and migration upon stimulation with atherogenic components such as angiotensin II.45
In migrating cells, the UPAR anchored to the cell membrane is focally redistributed to areas of adhesion at the apical part of the cells, where it interacts with different membrane integrins. It seems that the interaction between UPAR and integrins promotes cell actin cytoskeleton reorganization during migration processes.46 Recent studies by our group have shown that atherogenic concentrations of LDL induce changes in the subcellular localization of UPA and UPAR in migrating human vascular SMCs and their colocalization with actin,47 All together these changes might help to explain the mechanisms by which LDL inhibit human vascular SMCs migration kinetics.48
Modified LDL particles (enzymatic degradation) may favor in situ acute-phase inflammatory processes mediated by SMCs. In these cells, LDL particles induce synthesis of PTX3, a protein of the pentraxin family, to which C-reactive protein and serum amyloid P also belong,49 and thereby also contribute to the atherogenic process.
PLATELETS AND ATHEROSCLEROSIS
Cell components such as platelets and possibly progenitor cells also play a relevant role in the pathogenesis of atherosclerosis. Platelets are cells synthesized in bone marrow from megakaryocytes. They are devoid of DNA but contain messenger RNA derived from megakaryocytes and the transcription machinery necessary for protein synthesis.50 Platelets, in addition to their well known function in thrombosis and hemostasis, contribute to endothelial activation and modulate inflammatory responses, and so favor the onset and formation of atherosclerotic lesions and their subsequent thrombotic complications.51,52
Effect of Lipoproteins on Platelet Function
Changes in the composition and function of platelets in patients with hypercholesterolemia indicate that circulating lipoproteins affect platelet phenotype.53 Lipoproteins affect platelet function through binding to specific receptors (such as CD36, SR-B1, and LOX-1).54 However, so far, there is insufficient evidence that native LDL particles simply bind to the platelet surface or are internalized in intracellular vesicles through a process of endocytosis. Native LDL particles affect platelet function through activation of transduction signals or lipid exchange. Thus, native LDL particles can induce the synthesis or translocation of platelet membrane phospholipids or favor the insertion of phospholipids from the bloodstream and, as a result, alter the phospholipid composition of the platelet membrane.55 In contrast, binding between oxidized LDL particles and the platelet surface induces activation, morphological changes, and platelet aggregation, thereby contributing to the formation of thrombi, particularly after plaque rupture.56
Antithrombotic Properties of the Endothelium
Under physiological conditions, approximately 1.5×1012 platelets are continuously circulating throughout the arterial tree without any significant interactions with the vessel wall. The negative charge present on both platelets and vascular wall-related glycoproteins is responsible for an initial repellant effect that prevents interaction between platelets and vessel. In addition, there are 3 main pathways by which the vascular endothelium controls platelet reactivity: a) the arachidonic acid-prostacyclin pathway; b) the L-arginine-NO pathway; and c) the ecto-adenosine-diphosphatase pathway (ecto-ADPase or CD39).57 Endothelial cells are able to convert arachidonic acid into prostacyclin thanks to cyclooxygenase (COX) 1 or COX-2 and prostacyclin synthetase. Prostacyclin, in turn, inhibits platelet function by increasing intracellular cyclic AMP (cAMP).58 In fact, in our group, we have shown that selective COX-1 inhibition leads to vascular protection by the available synthesis of prostacyclin through vascular COX-2.59 With regard to the NO pathway, once synthesized by the endothelium from L-arginine, this molecule diffuses to the platelets and stimulates the production of cyclic GMP (cGMP). In turn, cGMP regulates cGMP-dependent protein kinases and causes a reduction in intracellular Ca2+ flux. Such decrease prevents GPIIb/IIIa conformational change (ie, activation) and hence decreases platelet-platelet interactions as described below.58 Finally, with regard to ecto-ADPase, this is an integral component of the endothelial surface and is activated according to the substrate concentration. It therefore limits the nucleotide plasma concentration (adenosine diphosphate [ADP] and adenosine triphosphate [ATP]). Thus, the activity of this enzyme slows the critical platelet recruitment and activation phase by removing nucleotides from the fluid medium.60
Along with these 3 pathways, the endothelium has other mechanisms for modulating thrombus formation by acting, mainly, on the blood coagulation pathway. These mechanisms include: a) the heparin sulfate pathway and thrombomodulin: both are integral components of the vascular endothelium and mediate the activation of antithrombin III and protein C, respectively, and thus block thrombin synthesis (potent platelet agonist), and b) synthesis and release of plasminogen tissue activator, which limits thrombus stabilization by promoting the degradation of the fibrin mesh.
Platelets and Endothelial Dysfunction
Endothelial dysfunction/activation in itself is able to induce adhesion of platelets to the vascular wall, without the need for exposure to deeper vessel surfaces (for example, vascular matrix) to the bloodstream. In fact, the availability of new in vitro research tools, such as intravital microscopy or genetically modified murine models for cardiovascular disease have helped to demonstrate that endothelial denudation is not an absolute requirement for platelet adhesion and subsequent activation to occur.61,62 It has been postulated that the molecular mechanism for platelet activation in the initial stages of atherosclerotic lesion comprise: a) a reduction in the mechanisms implicated in maintaining the antithrombotic properties; b) an increased generation of ROS associated with risk factors (hypertension, hypercholesterolemia, smoking, and diabetes correlate with a greater number of activated circulating platelets)63-66; and c) an increase in prothrombotic and proinflammatory mediators both in the bloodstream and immobilized on the endothelium.67 Thus, the endothelium is able to secrete abundant von Willebrand factor (vWf) in response to different inflammatory stimuli, and so promote platelet recruitment.
Platelet Adhesion and Activation
Currently, it is well established that the initial binding (rolling) of the platelets to the dysfunctional endothelium is mainly mediated by binding platelet GP Iba to endothelial vWF, as well as binding to the endothelial adhesion molecule P-selectin.61 In fact, just like vWF, P-selectin is found in both platelet alpha granules and in endothelial Weibel-Palade bodies,68 and upon cell activation it is rapidly translocated towards the cell surface, where it remains exposed for at least 1 hour.68 In fact, it is considered that P-selectin is an important marker of vascular disease, given that different studies have shown that platelets increase P-selectin expression on their surface as the formation of atherosclerotic lesions evolve.69 In addition to the expression of vWF and P-selectin by dysfunctional endothelial cells, the dysfunctional endothelium also expresses selectins, integrins (vascular cell adhesion molecule 1 [VCAM-1]), vitronectin anb3 receptor, and platelet endothelial cell adhesion molecule (PECAM-1), which favor adhesion of the platelets to the vascular wall.
After vascular damage, the dynamics of platelet deposition and the consequent thrombus formation is regulated locally by: a) the severity of stenosis; b) the type of lesion; and c) the composition of the atherosclerotic plaque.70,71 Fissure or rupture of an atherosclerotic plaque in the coronary arteries, whether spontaneous or induced (revascularization interventions), with the consequent thrombus formation, is essential for the development of acute ischemic syndromes, as has been satisfactorily demonstrated post-mortem in tissues from patients who died suddenly or shortly after an episode of unstable angina or myocardial infarction.72 In asymptomatic patients and those with stable angina, the organization of the thrombus is important in the progression of atherosclerosis. Rupture of the atherosclerotic plaque exposes components of the vascular matrix (eg, different types of collagen, vWF, fibronectin, laminin, fibulin, and thrombospondin)73 to the bloodstream, thereby favoring the interaction between platelets and the vascular wall. Plasma vWF is essential for thrombus formation at high shear rates (>650/s), typical in medium vessels or partially occluded large vessels. This is mainly because high flow rate conditions induce a conformational change in vWF that allows exposure of the A3 domains of its multimers to vascular collagen (type I and type III collagen in the deepest vascular layers and type VI collagen in the subendothelial layers). This binding induces, on the one hand, a conformational change in vWF in its A1 domain, which thereby allows interaction with the GPIba receptor of the GPIb/ IX/V platelet complex and, on the other, allows interaction of the Arg-Gly-Asp (RGD) domain of vWF with the GPIIb/ IIIa platelet receptor (integrin aIIbb3)74-76 (Figure 6). However, the high rate of dissociation between vWF and GPIb/V/IX indicate that these links cannot provide stable binding between platelets and the subendothelial matrix.77 Unlike GPIb/V/IX, the GPVI platelet receptor binds directly to collagen and induces an activation of other adhesion receptors such as integrins aIIbb3 (GPIIb/IIIa) and aIIb1.

Figure 6. Diagram illustrating the role of von Willebrand factor (vWF) in platelet adhesion. High flow rates induce conformational changes in vWF allowing interaction of its A3 domain with matrix collagen. This induces a conformational change in the A1 domain, thereby allowing interaction with glycoprotein (GP) platelet receptor Ib-IX-V. This interaction stimulates calcium release, subsequent platelet activation, and subsequent conformational change of the fibrinogen receptor (GPIIb/IIIa), which can interact with fibrinogen and vWF to favor the interaction between platelets (platelet aggregation process). ADP indicates adenosine diphosphate; RGD, Arg-Gly-Asp amino acid sequences.
Both aIIbb3 and aIIb1 act in unison to promote a firm, stable, and irreversible bond between platelets and the vascular endothelium,78,79 by direct binding with collagen (aIIb1) or with the C1 domain of vWF (aIIbb3).77
At low shear rates, platelet adhesion mainly occurs through the collagen receptor that directly binds to the platelet receptor GPIa-IIa (integrin a2b1).80 To a lesser extent, fibronectin, laminin, vitronectin, and thrombospondin also contribute to platelet adhesion by binding to GPIc-IIa, GP Ic'-IIa, vitronectin receptors, and to GPIV, respectively.
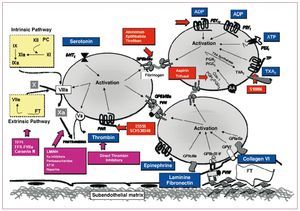
It should be highlighted that the presence of circulating agents such as epinephrine, thrombin, thromboxane A2 (TXA2), and ADP (released by hemolyzed red blood cells) also produces platelet activation through specific platelet receptors. In summary, collagen and vWF can be considered as primary agonists in the platelet activation-adhesion process. Additionally, ADP released by hemolyzed red blood cells in the area of the vascular lesion and the interaction with several circulating agonists with their platelet receptors favors and enhances this process (Figure 7).

Figure 7. Schematic representation of the main platelet activation pathways and therapeutic targets. AA indicates arachidonic acid; ADP, adenosine diphosphate; ATP, adenosine triphosphate; vWF, von Willebrand factor; PAR, protease-activated receptor; PG, prostaglandin; PKA, protein kinase A; TP, thromboxane receptor; TXA2, thromboxane A2.
Platelets and Monocytes/Macrophages
There is evidence that platelets, like red blood cells, penetrate the atherosclerotic plaque through rupture of angiogenic capillaries.81 In areas of endothelial damage, the close interaction between platelets and monocytes favors the differentiation of these cells into macrophages, and their transformation into mast cells. From in vitro studies, it has been shown that activated platelets can release cholesterol, which is probably internalized by SMCs and macrophages and stored in the form of lipid droplets.82 It has been reported that platelets also induce the formation of foam cells (CD68+) from progenitor cells positive for CD34. Recent evidence shows that this process can be prevented, at least partly, by HMG-CoA reductase inhibitors and peroxisome proliferator activated receptor agonists.83
An alternative pathway for the formation of mast cells could be phagocytosis of platelets by resident macrophages in the vascular intima, a theory put forward 40 years ago,84 and recently confirmed by in vitro studies showing that platelet phagocytosis by murine macrophages results in the formation of lipid-loaded macrophages.
LIPOPROTEINS AND ATHEROTHROMBOSIS
The mechanism involved in the sudden transformation of a stable vascular condition into a life-threatening event for the patient is usually the destabilization of the plaque with subsequent superposition of a thrombus.85 The risk of rupture of the atherosclerotic plaque depends more on its composition and vulnerability (type of plaque) than the severity of stenosis (plaque size). Different studies have revealed that more than 75% of myocardial infarctions occur in areas where coronary arteries have moderate stenosis (<50%).
Characteristics of Vulnerable Plaques
The predominant factors that affect the stability of atherosclerotic plaques are their cell composition and the ratio of ECM to lipid content. Unstable plaques and those with a high risk of rupture contain a substantial lipid core, rich in cholesterol esters and with little collagen, and a small number of SMCs. Inflammation within the fibrous cap, increased neovascularization, and fatigue constitute additional factors. Whereas SMCs account for 90%-95% of the cell component in initial lesions, this proportion decreases to 50% in advanced atherosclerotic plaques. These data reflect the importance of identifying and elucidating the cell mechanisms that lead to loss of SMCs in advanced lesions, as this might help to define new therapeutic strategies.
Atherogenic concentrations of LDL significantly reduce the migratory capacity of human vascular SMCs.48 Using proteomic techniques and confocal microscopy, our group has recently shown that LDL particles affect the expression and phenotypic profile of different proteins associated with the cytoskeleton of the SMCs, especially the myosin light chain in both the essential and regulatory isoforms, and also induce the phosphorylation of these proteins and a change in their subcellular localization.48 Phosphorylation of the myosin regulatory light chain (MRLC) is a key event for the formation of actin-myosin complexes during cell migration and the dynamics of actin fiber formation. This process is also regulated by proteins such as gelsolin and HSP27 chaperone, whose proteomic profiles are modified in SMCs exposed to atherogenic concentrations of LDL.87 In addition, the internalization of LDL by SMCs induces a decrease in MMP9 activity88 and may also potentiate the inhibitory effect of LDL on the migration of SMCs, thereby contributing to the instability and vulnerability of plaques in advanced stages.
Typically, plaques with a fine fibrous layer covering the lipid core and a high tissue factor (TF) content36 are those with greatest potential for rupture and induction of thrombosis.89 The proximity of TF and lipid-rich areas in advanced atherosclerotic lesions indicates that LDL particles are implicated in TF cell expression. In support of this hypothesis, it has been shown that modified lipoproteins affect the expression and activity of TF in SMCs. Schecter and colleagues90 have proposed that SMCs can secrete microparticles rich in TF through a mechanism that is independent of the cell apoptosis process. In fact, there is evidence of an increase in circulating TF-rich microparticles in patients with acute coronary syndromes and in patients with metabolic syndrome.91
Our group has shown that the interaction between LRP-1 and LDL aggregates is one of the mechanisms that induce TF expression in a process that depends on RhoA translocation to the membrane92 and the release of microparticles enriched in active TF to the ECM.93
Along with SMCs, macrophages are another important source of TF in the vessel wall. Expression of TF in monocytes appears to be regulated by PPARa.94 The differences in activation and secretion mechanisms of TF in SMCs and macrophages seem to depend on their distribution in the plasma membrane. Thus, TF and SMCs localize preferentially to the caveolae, whereas monocytes/ macrophages are mainly found in clathrin-enriched areas.95
PLATELET AGGREGATION AND ATHEROTHROMBOSIS
Once activated, platelets undergo a conformational change and release intraplatelet Ca2+. Such events are largely responsible for the subsequent platelet activation processes, including platelet degranulation, eicosanoid synthesis, expression of adhesion proteins, and exposure of a procoagulant platelet surface.
Platelet degranulation. This includes exocytosis of alpha, dense, and lysosomal granules content. ADP, serotonin, and calcium released from the dense granules are key elements in platelet function and the recruitment of new platelets.96,97 Specifically, ADP has an autocrine effect as it promotes stable platelet aggregation on interacting with ADP-specific receptors present on the platelet surface (P2Y1 and P2Y12) but also has a paracrine effect on binding to the ADP receptors on neighboring platelets, thereby amplifying the platelet activation process induced by other agonists. Release of growth factors of the alpha granules initiates vascular repair, and cytokines (alpha granules) and lysosomal enzymes (lysosomal granules) constitute the link with the immune response.
Eicosanoid synthesis. Phospholipase A2 initiates the activation of the arachidonic acid pathway, which is converted into TXA2 in a reaction catalyzed by COX-1 (to form prostaglandin G2/H2) and thromboxane synthetase (to form TXA2).98 TXA2 is released to the bloodstream, where it binds to TXA receptors (TP receptors) present on the surface of adjacent platelets, circulating inflammatory cells and on atherosclerotic plaque components amplifying and perpetuating the atherothrombotic process. In fact, we have demonstrated that blockade of TP receptors is associated with a marked reduction in the risk of thrombosis induced by intravascular stenting.99 In addition, TP receptor blockade has also shown to induce regression of vulnerable atherosclerotic plaques.100
Expression of adhesion proteins. Surface translocation of different adhesion proteins present on the platelet alpha granules (P-selectin, thrombospondin, GPIIb/IIIa, vWF, fibrinogen, fibronectin, and vitronectin) occurs, which amplify and perpetuate the platelet adhesion process and promote platelet interaction with white blood cells.
Exposure of a procoagulant platelet surface. A drastic change in the morphology of the platelet membrane occurs, from a smooth disc to a prickly sphere. This change favors the exposure, on the external membrane, of phospholipoprotein components rich in phosphatidylserine. This facilitates the binding and activation of the coagulation factors and subsequent thrombin formation (potent platelet agonist).101 In its turn, thrombin amplifies the initial activator effect of collagen and further promotes the activation, release, and recruitment of platelets. All together, such events induce the formation of eicosanoids and the appearance of fibrin filaments, initially in the exterior portion of the platelet thrombus, but also at the interstitial sites between adhered platelets. The binding of thrombin to fibrin or the artery wall masks thrombin receptors for antithrombin III, heparin, and cofactor II, such that the mural thrombus acts as a potent thrombogenic stimulus—dependent on thrombin and relatively resistant to heparin—that favors thrombus growth.102
Regardless of the trigger, platelet activation is regulated in the final part of the pathway by activation of the platelet GPIIb/IIIa receptor. The GPIIb/IIIa receptor is the most abundant protein on the platelet surface and is made up of 2 protein units (IIb and IIIa). Activation of this receptor supposes a conformational change in the 2 subunits such that the binding domain is exposed to a range of ligands. Fibrinogen (of plasma or platelet origin) is the protein that binds mainly to the binding domain of the GPIIb/IIIa binding receptor and its dimeric structure allows it to interact with 2 platelets at the same time, thereby favoring platelet aggregation. There are other ligands for GPIIb/IIIA that also participate, although to a lesser extent, in the interaction between platelets, such as vWF, fibronectin, vitronectin, and the CD40 ligand.103
In the final phase of thrombus formation, fibrinogen is converted to fibrin by thrombin, and this leads to stabilization of the platelet aggregates.77 In addition, activation of the signaling pathway occurs from outside in, thereby causing amplification of the initial signal and greater platelet activation and recruitment. This forms a mass that expands and continues to recruit more platelets as these reach the prothrombotic microenvironment. There is also recruitment of other blood cells; often red blood cells, neutrophils, and, at times, monocytes reach the lesion area intact, but respond to platelet secreted components.
ACTIVATION OF THE COAGULATION CASCADE
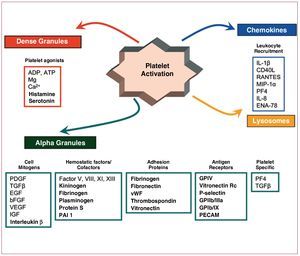
One of the early events after vascular rupture is activation of the coagulation cascade (Figure 8). Robust scientific evidence supports the fundamental role of TF as a potent inducer of the coagulation cascade, particularly TF expressed in foam cells present in the lipid core of atherosclerotic lesions.104-107 In addition to TF, both activated platelets and endothelial dysfunction have an important thrombogenic potential, promoting the coagulation cascade, with the subsequent formation of fibrin. In fact, dysfunctional endothelium changes its anticoagulant phenotype to a procoagulant state108 at the same time as platelets expose surface cofactors that can catalyze the formation of thrombin from prothrombin, the end product of the activation of the coagulation cascade and subsequent formation of thrombin, a potent platelet agonist and an important component in the pathogenesis of the atherothrombotic process. Thrombin signaling occurs through the 4 protease-activated receptors109 present on the surface of different atherosclerotic cell types. Specifically, PAR1 and PAR2 are present in SMCs and favor their proliferation; PAR1, PAR2, and PAR4 are present in macrophages and induce inflammatory pathways; and PAR1 and PAR4 are present in human platelets and their signaling culminates in the activation of the fibrinogen receptor.

Figure 8. Schematic of platelet components implicated in the coagulation cascade and the atherosclerotic process. ADP indicates adenosine diphosphate; bFGF, fibroblast growth factor; EGF, endothelial growth factor; ENA, neutrophil-derived peptide activator of epithelial cells; GP, lycoproteins; IGF, insulin-like growth factor; IL, interleukin; MIP-1, macrophage inflammation protein 1; PAI-1, plasminogen activator inhibitor 1; PDGF, platelet-derived growth factor; PECAM, platelet and endothelial cell adhesion molecule; PF4, platelet factor 4; TGFb, transforming growth factor b; VEGF, vascular endothelial growth factor; vWF, von Willebrand factor.
Therefore, thrombin, through PARs, influences a wide variety of responses that favor the development of atherosclerotic lesions.110 Activation of these receptors is brought about by means of a proteolytic process; that is, binding of thrombin with PAR leads to excision of the amino-terminal segment of the receptor that generates new amino acid terminal chains (SFLLRN for PAR1 and GYPGKF for PAR4), which subsequently stimulate other receptor regions.111
In summary, the fibrinogen receptor is not only activated through the endothelial receptors or collagen GPIb/IX/V and GPVI, but also through receptors coupled to G proteins such as thrombin (PAR1, PAR4) or the ADP receptors (P2Y1, P2Y12), which enhance GPIIb/IIIa-dependent platelet aggregation, as well as the subsequent thrombus formation.
PLATELET-DERIVED MICROPARTICLES (MPps)
A range of evidence in recent years suggests that the role of platelets in atherosclerosis and its thrombotic complications may be mediated, in part, by secretion of MPps (microvesicular platelets formed during the platelet activation process).112,113
High concentrations of circulating MPps have been reported in patients with atherosclerosis, acute vascular syndromes, and/or diabetes mellitus,112 suggesting a potential correlation between the quantity of microparticles (MPs) and the clinical severity of the atherosclerotic disease.114
Although blood contains MPs formed from different cell types (leukocytes, red blood cells, and endothelial vascular cells and smooth muscle cells), most circulating MPs are of platelet origin. MPs contain a protein load derived from the stem cells from which they originate, with proteins from both the plasmatic membrane and the cytoplasmic region situated close to the part of the cells where they were formed. Therefore, on their surface, MPps present a multitude of platelet adhesion receptors and chemokines (such as P-selectin, GPIIb/IIa, and GPIba, among others), which not only induce cytokine production by circulating monocytes and the endothelium,115 but also increase aggregation and recruitment of leukocytes by means of P-selectin-dependent interactions and their PSGL-1 ligand.116 Similarly, MPps can adhere to the activated endothelium, thereby enhancing leukocyte adhesion through an increase in the expression of the adhesion molecule ICAM-1117 and increasing the inflammatory environment through the production of interleukins (IL-1, IL-6, and IL-8).118 Moreover, it has been reported that circulating MPps could serve as cell transfer modules for a range of proinflammatory mediators and platelet receptors.119 Thus, the high concentrations of MPp not only reflect a platelet activation epiphenomenon, but should also be considered as a transport and transcellular release system. Ex vivo generated MPps lead to in vivo vascular contraction, as they induce a local increase in TXA2concentration.120 Although circulating MPs currently have a limited potential prognosis, the results available until now indicate that the characterization of their origin and their protein composition may be a valuable tool for determining cardiovascular risk, even in patients without clinical manifestations.
LIPID-LOWERING TREATMENTS AND RISK OF THROMBOSIS
Different clinical trials have shown the importance of lowering LDL-cholesterol (LDL-C) through the administration of 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors (statins) for primary and secondary prevention of cardiovascular disease,121,122 and this is also true for patients with heterozygotic familial hypercholesterolemia.121-123 The results obtained in the ASTEROID clinical trial (A Study to Evaluate the Effect of Rosuvastatin on Intravascular Ultrasound-Derived Coronary Atheroma Burden) have shown, through intravascular ultrasound analysis (IVUS), that aggressive lipid-lowering treatment can induce regression of already established coronary atherosclerotic plaques.124 Moreover, a multicenter study currently underway will evaluate whether such regression is associated with changes in the atheromatous plaque composition towards a greater stabilization through IVUS combined with virtual histology.125 Nevertheless, some clinical trials show that the benefits derived from statin therapy are largely independent of the baseline reduction in LDL-C, and this raises the possibility of clinically beneficial effects beyond those arising from reduction in LDL cholesterol.126 In fact, it has been shown that statins have antiinflammatory, antioxidant, and antithrombotic properties.127-130 In this context, we have demonstrated that cholesterol levels regulate platelet reactivity.130 Moreover, preliminary studies conducted in hypercholesterolemic patients with stable coronary artery disease have shown that treatment with pravastatin is associated with a marked reduction in thrombus formation and an improved fibrinolytic profile.131 In the same context, we have shown a notable and persistent improvement in endothelial function in patients with familial hypercholesterolemia after simvastatin therapy .132 The underlying mechanisms for these thromboprotective effects have yet to be elucidated, although we have performed in vitro studies130 and in vivo studies133,134 that link a lower platelet reactivity with a reduction in the activation of RhoA (a small protein of the Rho GTPase family implicated in the reorganization of the platelet cytoskeleton), as well as a lower TF expression in the vascular wall.133,134 The relevance of HMGCoA reductase inhibition for normalizing and stabilizing the cardiovascular system is reflected in 2 recent scientific papers. In an experimental model very similar to humans, it has been shown that the administration of rosuvastatin after ischemic reperfusion reduces myocardial damage and improves myocardial contractility.135 On the other hand, the Efficacy of Atorvastatin Reload in Patients on Chronic Statin Therapy Undergoing Percutaneous Coronary Intervention (ARMYDARECAPTURE) study has shown that a loading dose of atorvastatin before coronary revascularization (80 mg 12 hours prior to and 40 mg at the beginning of the procedure) in patients already treated with statins improves the resulting clinical events.136
ABBREVIATIONS
ADP: adenosine diphosphate
AMP: adenosine monophosphate
EC: endothelial cell
ECM: extracellular matrix
GP: glycoprotein
LDL: low-density lipoprotein
LRP-1: low-density lipoprotein receptor-related protein 1
MPp: platelet microparticle
PAR: protease-activated receptor
SMC: smooth muscle cell
TF: tissue factor
UPA: urokinase plasminogen activator
vWF: von Willebrand factor
Section sponsored by the Laboratorio Dr. Esteve
This article has been possible thanks to the funding of CIBERobn CB06/03, SAF 2006-10091, FIS-PI071070, REDINSCOR RD06/0003/0015 and the Fundación de Investigación Cardiovascular. Dra. Gemma Vilahur is contracted Ramón y Cajal (Ministerio de Ciencia e Innovación).
Correspondence: Prof. L. Badimón.
Centro Investigación Cardiovascular.
Sant Antoni Maria Claret, 167. 08025 Barcelona. España.
E-mail: lbadimon@csic-iccc.santpau.es