Arrhythmogenic cardiomyopathy (AC) is a hereditary heart muscle disease disorder consisting of progressive fibrofatty replacement of right ventricle (RV) and left ventricle (LV) myocardium, leading to delayed electrical conduction and malignant ventricular arrhythmias. It represents one of the major etiologies of sudden cardiac death (SCD) in young and apparently healthy individuals. At the cellular level, significant advances have been made in terms of discoveries regarding genetic and pathophysiological mechanisms in the last few years, specifically with nondesmosomal genetical targets with LV involvement. In the next generation sequencing era, more than 15 genes have been involved in the etiopathogenesis of AC, with a common final pathological pathway of cell death and fibrosis.1 Indeed, the current 2010 Task Force diagnostic criteria include the presence of a pathogenic mutation categorized as associated or probably associated with AC.2 Most classic AC show predilection for mutations directly affecting desmosomal proteins, but genetics have helped to identify a much broader spectrum of the disease, extending its genetic spectrum beyond the desmosomal genes. Examples of nondesmosomal genes associated with clinical and key pathological findings of predominantly LV-AC are genes encoding proteins from the cytoskeleton (DES), the nuclear envelope (LMNA, TMEM43), the sarcoplasmic reticulum (PLN), and the Z-disc (FLNC).1
The latter bring us to the question of how a molecule from the nuclear envelope or the cytoskeleton lead to an AC phenotype with RV, or mainly LV or biventricular involvement with abnormal electrical behavior. Cardiomyocytes form structural and electrical connections via desmosomes, adherens and gap junctions, which all occur in mixed-type junctions, known as area composita, located at the intercalated disc. One important function of desmosomes is to connect adjacent cells mechanically by joining their intermediate filaments to create a unified cytoskeletal network. Through these interactions, desmin coordinates movement of neighbouring Z-discs with the nuclear and plasma membranes.3 Consequently, this mechanical network is tightly related to filamin C at Z-discs and the LINC (linker of nucleoskeleton and cytoskeleton) complex composed by lamins and LUMA (TMEM43) proteins, as well as ion plasma membrane channels such as the sodium channel NaV1.5, among others. Thus, defects in this network might alter the structural integrity and mechanotransduction of cardiomyocytes, mirroring a proposed mechanism of desmosome mutations in AC. There are still large gaps in our knowledge of the molecular pathogenesis of this condition but it is possible that the AC phenotype is the common expression in a failing cascade in this macromolecular complex from sarcolemma and area composita to nuclei.
The diagnosis of AC is challenging and relies on the combination of electrical, structural, genetic and pathological data, as well as family history. Task Force diagnostic criteria, first published in 1994 and then updated in 2010,2 combine all these data together, requiring, for a definite AC diagnosis, 2 major, 1 major and 2 minor or 4 minor criteria. AC definite diagnosis is particularly difficult due to a characteristic variable disease penetrance and expression, even among family members with the same mutation, with a relatively common incidence of silent gene mutation carriers. To detect subtle myocardial changes suggestive of AC, cardiac imaging has rapidly evolved in the last decade. Thus, advanced cardiac imaging should be one of the cornerstones of AC diagnosis, with cardiac magnetic resonance (CMR) being the most sensitive technique, not only for cardiac structure definition, but also for tissue characterization.
CMR is gaining importance in the field of ischemic and nonischemic cardiomyopathy, either for enhanced disease characterization, providing key points for differential diagnosis, as well as to improve prognostic risk stratification.4 Of particular interest is the diagnostic value of CMR in other nonischemic cardiomyopathies, as specific late gadolinium enhancement (LGE) patterns have been proposed for several clinical scenarios.5 Furthermore, the extent of LGE seems to correlate well with adverse arrhythmic events, including SCD, in dilated and hypertrophic cardiomyopathies. However, robust data about the role of CMR in left-dominant AC are still lacking.
In a recent article published in Revista Española de Cardiología, Feliu et al.6 present an elegant study, which endeavors to delve into the knowledge of the diagnostic and prognostic value of CMR in left dominant AC. The authors should be congratulated for a brilliant work that helps to highlight the role of CMR in AC. In a sufficient patient sample size, they were able to establish a correlation between the presence of extensive LV-LGE and poor outcome due either to arrhythmic or heart failure major events. Moreover, they provide evidence of a common characteristic CMR finding, which they call “the rat-bite sign”, in a significant proportion of the cases, as a result of LV fatty infiltration (FI), highly suggestive of AC. Finally, their results confirm the previously described inferolateral-subepicardial and circumferential LGE pattern as a typical AC diagnostic clue.
From the clinical perspective, this sample is likely overrepresented with clinically advanced cases. Indeed, 80% of the included cases are probands, and most of them after an episode of sustained ventricular arrhythmia, with an automatic defibrillator implanted for secondary prevention of SCD. This may explain the relatively high rate of adverse events observed. In addition, the mixed nature of the study design, taking into account the initial arrhythmic cardiac event for the follow-up statistical analysis, adds some bias to the interpretation of the real clinical outcome. In larger prospective samples, such as that of Mazzanti,7 the rate of adverse cardiac events at 5 years was significantly lower. This ambispective nature of sample of patients in the study by Feliu et al. may limit the conclusions about the high prevalence of CMR findings such as the “rat-bite sign” or the extensive inferolateral LGE, which might not be present in relatives and mutation carriers in a more preclinical phase. This still represents a central challenge in AC diagnosis in order to achieve an early diagnosis.
We agree with Feliu et al.6 that LV dominant and biventricular forms of AC are underrepresented in the CMR 2010 Task Force criteria2 and a future update of these criteria should include both phenotypes. It is well recognized that LV involvement is common in AC. In keeping with previous data, we have also reported either regional wall motion abnormalities or LGE in up to 79.5% of cases,8 and recent contributions have shown that up to 70% of SCD in AC autopsies showed histopathologic biventricular involvement.9 In the last few years, there has been growing interest in this LV phenotype. In the absence of revised criteria, recent CMR studies have shown some features that might help to understand left-dominant and biventricular forms. Initially, Sen-Chowdhry et al.10 proposed as diagnostic traits the presence of LV aneurysms, mild LV dilation, systolic impairment and LGE with subepicardial/midmyocardial distribution. Certainly, LGE LV involvement in the literature is described in subepicardial/midwall layers and has been correlated histopathologically showing fibrofatty infiltration with a characteristic gradient from the outer to the inner stratum (“wave-front phenomenon”), concerning subepicardial layers with some extension to mesocardium.11
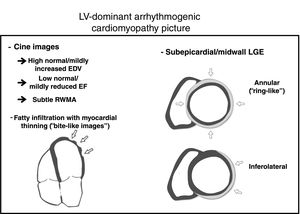
The 2010 Task Force criteria acknowledge that fibrofatty replacement,2 and not only FI, should be the true hallmark of the disease (figure 1). Fatty suppression CMR sequences has several limitations such as high interobserver variability, difficulty in the interpretation, and low specificity. It is important to be aware that FI can be observed in other clinical scenarios. RV-FI can be found in up to 50% of RV of normal hearts in the elderly with myocytes appearing to be pushed apart rather than replaced, with no evidence of fibrosis, myocyte degeneration, or inflammation.12 Moreover, an increase of fatty tissue in the subepicardium is a regular finding in the obese (adipositas cordis) and should not be misinterpreted as AC. Finally, FI can be observed in other entities such as healed myocardial infarction, cardiac lipoma, tuberous sclerosis, and dilated cardiomyopathy.13 In AC, FI has been previously described both in RV and LV, as “fingerlike” projections disrupting the normal wall contour, being preferential locations epicardial RV and LV free walls,13 which are in keeping with the findings of Feliu et al.6 The novel “rat-bite sign” described by these authors is a suggestive and valuable finding to strongly suspect underlying AC. However, the absence of FI in some disease forms, such as Carvajal syndrome (which frequently associates LGE), reduces its sensitivity as a diagnostic marker of AC.14

The wide spectrum of the disease can be expressed in different ways and underlying genetic status might play an important role. In fact, we have observed that patients with desmosomal mutation genes mainly manifest as typical arrhythmogenic right ventricular cardiomyopathy with or without biventricular (BV) phenotype, while nondesmosomal mutation genes express as LV and BV forms.8 Recent studies have shown that not only the presence of LGE, but also its extent and subepicardial inferolateral location are important risk prognostic factors.5 We believe that LGE distribution depends on the genetic condition, since we observed that FLNC mutation carriers show a predilection for the inferolateral wall, while desmin mutation had a distinctive and extensive subepicardial annular (ring-like pattern).8 Similar to FI, these LGE patterns, although not pathognomonic, are highly suggestive of AC. Therefore, LGE, as a hallmark of fibrosis replacement, should to play a central role in the forthcoming AC criteria, but clues such as FI with myocardial thinning, slightly increased LV volumes, mildly reduced ejection fraction or subtle regional wall motion abnormalities help to support the final diagnosis (figure 1).
Finally, Feliu et al. have identified some clinical risk stratification predictors of poor outcome, such as male sex and sports practice.6 Sex has been proposed as an arrhythmic risk marker in several gene-specific subtypes of AC such as LMNA, FLNC, TMEM43, and DES.15 Previous studies in a more heterogeneous sample of AC/ARVC patients have also found a positive link between male sex and poor outcome and the data of Feliu et al. serve to confirm this finding. Regarding sports, although this work was not intended for this specific aim, the results remain controversial. Previous studies have clearly demonstrated that arrhythmic risk is directly related to previous competitive sports practice. However, most of the studies in the field are not completely accurate in terms of physical activity recording, and, to date, specific advice about sports should not categorically recommend the avoidance of any kind of sports practice.
In summary, we agree with the authors that it is time to update the diagnostic criteria of AC. There are emerging data from myocardial tissue characterization and LGE patterns by cardiovascular imaging, but also a subset of high-risk genotypes with well-known predilection for the LV, specifically the inferolateral subepicardium. Worldwide cardiologists expect an early revision of the diagnostic criteria integrating all these advances leading to appropriate management strategies.
CONFLICTS OF INTERESTNone declared.