To the Editor,
.
A 45-year-old man was referred to our department for evaluation of recurrent syncope. The episodes were first observed 1.5 years ago and had recently increased in frequency. They were preceded by dizziness and vertigo and sometimes led to minor injuries. The patient also reported substantial weight loss, night sweats, and diarrhea. During previous hospital stays neurologic disorders like seizures and multiple sclerosis, as well as borreliosis and Guillain-Barré syndrome, had been ruled out. He also had tested negative for celiac disease. A dual-chamber pacemaker had been implanted for suspected chronotropic incompetence but this did not relieve the symptoms.
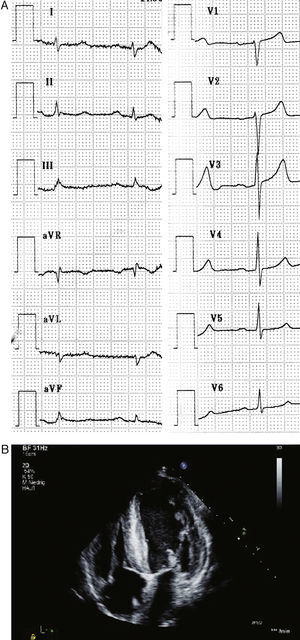
Physical examination revealed a cachectic patient with a systolic ejection murmur without radiation, bilateral weak deep tendon reflexes and hypesthesia in a symmetrical glove and stocking pattern. The patient had wasting of the small hand muscles and walking on toes or heels was impaired. Orthostatic testing revealed postural hypotension. Routine laboratory results were normal. A 12-channel electrocardiogram (ECG) demonstrated peripheral low voltage (Figure 1A). Holter ambulatory monitoring displayed normofrequent sinus rhythm without any arrhythmia. Pacemaker telemetry detected 0% stimulation at a minimum stimulation rate of 50 bpm and no arrhythmic episodes in the diagnostic memory. However, 24-h blood pressure monitoring revealed hypotensive episodes with a mean of 81/44mmHg. Transthoracic echocardiography showed massive biventricular hypertrophy compatible with infiltrative disease and thickened mitral and tricuspid valve without significant dysfunction (Figure 1B). Left ventricular ejection fraction was normal (65%). Hypertrophic obstructive cardiomyopathy was ruled out by absence of intraventricular pressure gradient or systolic anterior movement of the mitral valve at rest and under provocation tests. Motor and sensory nerve conduction study confirmed axonal polyneuropathy.

Figure 1. A: Electrocardiogram showing sinus rhythm and peripheral low voltage. B: Transthoracic echocardiogram showing hypertrophic left ventricular wall and septum and thickened mitral and tricuspid valve.
The combination of polyneuropathy and infiltrative heart disease with peripheral low voltage in ECG aroused suspicion for systemic amyloidosis with cardiac involvement.1 We performed a rectal biopsy in our patient, which stained positive with Congo red and demonstrated apple-green birefringence under polarized light microscopy. Serum electrophoresis showed normal transthyretin (TTR) as well as a basic TTR mutant. Molecular analysis of TTR-gene confirmed heterozygosity for a point mutation at exon 2 (TTR-G27.A); thus the diagnosis of familial amyloidosis of the TTR-type was confirmed. The medical records of the patient's deceased mother were obtained, and indicated that she had suffered from symptoms compatible with amyloidosis.
TTR is a transport protein for thyroxin and is primarily synthesized in the liver. Amyloidogen mutations of the TTR-gene (ATTR) are the most common causes of familial amyloidoses.2 They constitute a group of autosomal dominant diseases in which a mutant protein leads to deposition of amyloid protein in multiple organs. Since they predominantly affect the peripheral and autonomous nervous system, they are also denominated familial amyloid polyneuropathies (FAP). Involvement of the cardiovascular system can mimic hypertrophic cardiomyopathy, as it did in our patient at first sight.3 Recurrent episodes of syncope as an isolated feature of FAP have been described. They most likely result from orthostatic dysfunction caused by autonomic neuropathy.4
Our patient was found to be eligible for liver transplantation as the only definitive therapy for ATTR. His replaced liver was used as a donor organ for transplantation into another patient with terminal liver disease. This procedure is referred to as “domino transplantation.”5 Since it usually takes 3 decades for familial amyloidosis to manifest, this can safely be done in older recipients.6 After transplantation, clinical improvement of neuropathy and regression of amyloid deposits in the organ tissues have been reported.2
This report illustrates a rare cause for recurrent syncope, which is a common presenting problem in a cardiovascular clinic. The triad of peripheral neuropathy, peripheral low voltage, and infiltrative myocardial disease indicated systemic amyloidosis with cardiac involvement.
Corresponding author: markus.linhart@ukb.uni-bonn.de